Abstract
Einleitung: Kongenitale Anophthalmie ist das klinische Fehlen des Auges bei der Geburt. Sie resultiert aus der fehlenden Entwicklung oder Regression der primären Sehnervenblase während des Embryonallebens. Er ist selten und kann isoliert oder in Verbindung mit anderen okulären oder allgemeinen Geburtsfehlern auftreten.
Beobachtungen: Wir berichten über drei Kinder aus derselben Familie im Alter zwischen 4 Monaten und 8 Jahren mit beidseitigem kongenitalem Anophthalmus. Ihre Anamnese ist gekennzeichnet durch einen Mangel an Beratung und pränatalen Laboruntersuchungen. Die ophthalmologische Untersuchung objektivierte, das Öffnen der Augenlider mit zwei Augen, eine Füllung des Hohlraums durch das Bindehautgewebe in 3 Fällen, was auf einen bilateralen Anophthalmus hindeutet, und bestätigte den Augenultraschall.
Diskussion: Der kongenitale Anophthalmus ist eine seltene Fehlbildung. Sie kann isoliert oder in ein Fehlbildungssyndrom integriert sein. Das klinische Bild ist meist unilateral. Die Diagnose wird hauptsächlich klinisch gestellt und durch Augenultraschall bestätigt. Die Ursachen sind vielfältig und umfassen Chromosomenaberrationen, genetische Mutationen, Vergiftungen und während der Schwangerschaft erworbene Infektionen.
Fazit: Die Entdeckung einer kongenitalen Anophthalmie erfordert eine umfassende Untersuchung, um die Ätiologie zu ermitteln. Auch die psychologische Unterstützung der Eltern bleibt ein wichtiger Schwerpunkt.
Schlüsselwörter
Anophthalmus, kongenital, Augenultraschall, Chromosomenaberrationen
Einführung
Anophthalmus ist das klinische Fehlen des Auges. Es handelt sich um eine extrem seltene Augenanomalie. Es gibt zahlreiche Ursachen wie Chromosomenanomalien, genetische Mutationen, Vergiftungen und Infektionen. Der angeborene Anophthalmus kann isoliert oder in Verbindung mit schweren polymorformativen Syndromen auftreten. Sie ist einerseits verantwortlich für einen Mikroorbit und eine homolaterale kraniofaziale Hemiatrophie und andererseits für eine schlechte Entwicklung der Anhängsel, die Probleme bei der prothetischen Anpassung verursachen. Die Diagnose konnte durch pränatale Bildgebung verbessert werden, und die therapeutische Behandlung umfasst im Wesentlichen Orbitalprothesen. Wir berichten über 3 Fälle von beidseitigem kongenitalem Anophthalmus in derselben Familie.
Fall 1
Es handelt sich um ein männliches Kind im Alter von 8 Jahren, das bei der Geburt geboren wurde und wegen einer beidseitigen Abnahme der Sehschärfe konsultiert wurde. Die biologische und zerebrale CT-Untersuchung vor der Geburt war nicht möglich. Die klinische ophthalmologische Untersuchung ergab ein Fehlen der Lichtwahrnehmung in beiden Augen; beidseitiger Anophthalmus; die Anhängsel sind vorhanden und normal und bei der Öffnung der Augenlider füllt eine Bindehautstruktur die gesamte Orbitalhöhle ohne Augapfel aus. Die angeforderte Computertomographie der Augenhöhle und des Gehirns bestätigte das Fehlen des Augapfels mit dem Vorhandensein von Fettresten, Augenmuskeln und einer Atrophie des Sehnervs in der Augenhöhle (Abbildung 1). Die Gesamtbilanz war normal. Eine Magnetresonanztomographie konnte angesichts der finanziellen Schwierigkeiten der Eltern sowie der genetischen Bilanz (Karyotypisierung) nicht durchgeführt werden. Eine chirurgische Behandlung mit Einsetzen einer Prothese wurde vorgeschlagen, aber mangels einer geeigneten technischen Plattform nicht durchgeführt. (Abbildung 2)
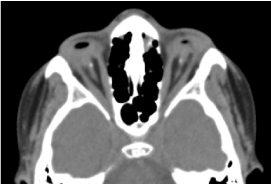
Abbildung.1. CT-Aufnahme der Augenhöhle: Fehlen des Augapfels mit Restfett, Muskeln und einer Atrophie des Kalibers des Nervus otocervalis in der Augenhöhle.

Abbildung. 2. 8-jähriges Kind mit beidseitigem Anophthalmus beim Öffnen der Augenlider
Fall 2
Es handelte sich um ein 8-jähriges männliches Kind, bei dem keine pränatale Untersuchung (biologisch und ultraschall-echographisch) durchgeführt wurde. Er wurde wegen einer beidseitigen Abnahme der Sehschärfe in die augenärztliche Sprechstunde gebracht. Die klinische ophthalmologische Untersuchung ergab ein Fehlen der Lichtwahrnehmung in beiden Augen, einen beidseitigen Anophthalmus, Anhängsel ohne Besonderheiten und beim Öffnen der Augenlider eine den Hohlraum füllende Bindehautstruktur). Die angeforderte Computertomographie der Augenhöhle und des Gehirns bestätigte das Fehlen des Augapfels mit dem Vorhandensein von Fettresten, Augenmuskeln und einer Atrophie des Sehnervenkopfes in der Augenhöhle (Abbildung 3). Die allgemeine Untersuchung des Kindes war normal. Die Magnetresonanztomographie und der Karyotyp konnten aus denselben Gründen wie zuvor erwähnt nicht durchgeführt werden, ebenso wenig wie die medizinische Behandlung. (Abbildung 4)
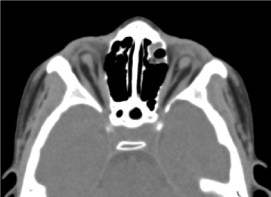
Abbildung.3Fehlen des Augapfels mit Restfett, Muskeln und einer Atrophie des Kalibers des N. otocervalis in der Orbitalhöhle.
Fall 3
Es handelt sich um einen 4 Monate alten weiblichen Säugling, der bei der Geburt geboren wurde und eine gute psychomotorische Entwicklung und ein gutes Körpergewicht aufwies und wegen einer beidseitigen Abnahme der Sehschärfe und eines permanenten Lidschlusses seit der Geburt in die augenärztliche Sprechstunde kam. In der Anamnese des Säuglings finden sich zwei pränatale Konsultationen, aber keine pränatalen biologischen Untersuchungen und Ultraschalluntersuchungen. Die klinische ophthalmologische Untersuchung ergab ein Fehlen der Lichtspur; bilateraler Anophthalmus; normale Aspekte der Anhänge und der Öffnung der Augenlider, ein leerer Augapfel, der ein mikrophthalmisches Erscheinungsbild ergibt, die Details der Iris sind nicht analysierbar und ein Fehlen einer vorderen Augenkammer. Die pädiatrische Konsultation auf der Suche nach anderen Fehlbildungen ergab keine Besonderheiten auf klinischer Ebene.
Die angeforderte Orbitazerebraltomographie bestätigte das Fehlen eines Augapfels mit dem Vorhandensein von Restfett, Augenmuskeln und einer Atrophie des Kalibers des Sehnervs in der Orbitalhöhle (Abbildung 5). Die Magnetresonanztomographie und die Karyotypisierung konnten nicht durchgeführt werden; ebenso wenig wie seine medizinische Behandlung (Abbildung 6).

Abbildung 4. 6-jähriges Kind mit beidseitigem Anophthalmus beim Öffnen der Augenlider.
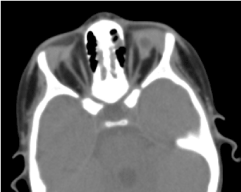
Abbildung 5. Fehlender Augapfel mit Restbläschen, Muskeln und einer Atrophie des Nervus otocervalis in der Augenhöhle.

Abbildung 6.. 4 Monate altes Kind; beim Öffnen der Augenlider konnte man, besonders am rechten Auge, eine Skizze der Hornhaut erkennen, aber die Iris ist nicht individualisierbar.
Diskussion
Kongenitale Anophthalmie oder klinisches Fehlen des Auges bei der Geburt ist eine seltene Fehlbildung . Ihre Inzidenz ist variabel. Nach Schätzungen von Mouriaux und Roth liegt sie bei weniger als einem Fall pro 10000 Geburten. Nach einer spanischen Untersuchung liegt sie bei 21,34 pro 100 000 Geburten und nach einer britischen Studie bei 10 pro 100 000 Geburten. Bei Aranjo liegt die Inzidenz bei 0,6 pro 10000 lebend geborenen Kindern. Dieser Unterschied in der Inzidenz erklärt sich durch die Variabilität des Auftretens dieser Fehlbildung. Die Häufigkeit der Anophthalmie wird nicht durch Rasse oder Geschlecht beeinflusst. In einem Drittel der Fälle kann Anophthalmus isoliert oder in ein polymalformatives Syndrom integriert sein. Diese Beobachtung deckt sich mit denen mehrerer Autoren. So können bei Verna in etwa einem Drittel der Fälle sowohl Anophthalmus als auch Mikrophthalmus isoliert auftreten oder in ein Patau-Syndrom (Trisomie 13) oder andere Syndrome integriert sein. Kouassi berichtete auch über einen Fall von beidseitigem kongenitalem Anophthalmus im Rahmen des Patau-Syndroms. Ebenso Diomandé in Bouaké, der über einen Fall von echtem kongenitalem Anophthalmus in Verbindung mit schwerem Mikrophthalmus bei einem Neugeborenen ohne andere beobachtete klinische Anomalien berichtete.
Außerdem stellte Stoll fest, dass 90 % der Anophthalmusfälle mit Fehlbildungen einhergehen. Daher ist es wichtig, bei Vorliegen einer kongenitalen Anophthalmie oder eines extremen Mikrophthalmus eine gründliche pädiatrische Untersuchung durchzuführen, um nach begleitenden Anomalien zu suchen, die manchmal schwerwiegend sein können, wie z. B. eine Anenzephalie oder eine Agenesie des gesamten Sehnervenkopfes. Der klinische Anophthalmus ist selten beidseitig. Moderne paraklinische Untersuchungen wie die Computertomographie und die Magnetresonanztomographie ermöglichen es, die damit verbundenen Fehlbildungen zu untersuchen. Nach Speeg-Schatz gibt es drei Arten von kongenitalem Anophthalmus: primäre Anophthalmitie, sekundärer Anophthalmus und konsekutiver sekundärer Anophthalmus als Folge der fehlenden Einstülpung der Fossa optica, des vollständigen Stillstands der Entwicklung des vorderen Neuralrohrs oder der Degeneration der Sehnervenblase nach ihrer Einstülpung. Für Stricker kann die kongenitale Anophthalmitie nach zwei Modi analysiert und klassifiziert werden.
Aus anatomischer Sicht müsste man zwischen echtem Anophthalmus und Embryopathien unterscheiden, die durch das Fehlen jeglicher Augenkontur gekennzeichnet sind. Dies kann nur durch eine histologische Untersuchung bestätigt werden.
Klinisch gesehen muss der klinische Anophthalmus oder der extreme Mikrophthalmus von anderen, moderateren Mikrophthalmen mit manchmal besserer funktioneller Prognose unterschieden werden. In den letzteren Fällen gibt es einen mehr oder rudimentären Augapfel (wie in unseren 3. klinischen Fällen), der manchmal von einer kolobomatösen Zyste begleitet wird, die als physiologische expansive Prothese fungieren kann. Klinisch gibt es keine Augenstruktur, aber ein Augenultraschall, eine Computertomographie und eine Magnetresonanztomographie würden das Vorhandensein einer Skizze des Augapfels und des Sehnervs zeigen.
Morax stellte jedoch fest, dass eine echte Anophthalmie nicht existiert. Ihm zufolge würde die Neurobildgebung in den ausgeprägtesten klinischen Fällen das Vorhandensein eines Sehnervs und einer winzigen Augenskizze zeigen. In unseren ersten beiden klinischen Fällen zeigte der Augenultraschall das Fehlen einer okulären Rauigkeit. In Ermangelung von CT- und MRT-Ergebnissen können wir jedoch nicht formell zu dem Schluss kommen, dass kein Sehnerv vorhanden ist.Die Ätiologie des Anophthalmus ist komplex und vielfältig. Sie kann durch Chromosomenanomalien, insbesondere Trisomie 13 oder Trisomie 18, vererbt werden. In diesem Fall handelt es sich um Anophthalmus mit zerebralen Fehlbildungen oder in Verbindung mit einer Dysplasie der Medianuslinie. Andere genetische Ursachen sind Chromosomenmutationen, die im Rahmen von Chromosomen-Deletionen oder unabhängig von einer Aberration des Karyotyps auftreten können. SOX2-Anomalien sind die wichtigsten davon. Fantes diagnostizierte eine Mikrodeletion in Verbindung mit einer Translokation t (3; 11) (q26-3 p11-2) und einer Mutation des SOX2-Gens bei einem Patienten mit beidseitigem Anophthalmus. Es wird angenommen, dass die Haploinsuffizienz von SOX2 für Anophthalmus, Mikrophthalmus, septo-optische Dysplasie und eine Störung der Entwicklung des vorderen Gehirns verantwortlich ist. Ye merkte an, dass der SOX2-Defekt eine multiple Störung des organischen Systems einschließlich Anophthalmus und Mikrophthalmus verursacht. Andere Gene wie PAX6, OTX2, CHV10, RAX2, SIX6, NF1, TWIST, SHH, BMP4 und BMP7 seien am Auftreten von Anophthalmie beteiligt. Laut Schilter würden weniger als 40 % der bekannten genetischen Mutationen die Anophthalmie erklären. Für ihn wäre eine Mutation der Gene NF1, PAX6, SOX2 für Anophthalmie oder Mikrophthalmus verantwortlich. Sie haben eine Mutation des Gens OTX2 beschrieben, die in einer chinesischen Familie mit Anophthalmie und kongenitalem Mikrophthalmus assoziiert ist. Veronica beobachtete bei einem Patienten eine Mutation des RAX2-Gens, die zu einseitigem Anophthalmus führte. Mutationen im TWIST-Gen bei Chromosom-7-Deletionen können zu einer Gesichtsdysgenesie mit Anophthalmus führen. Gallardo wies die Mutation eines Allels des Gens SIX6 bei einem Patienten mit beidseitigem Anophthalmus nach.
Ohne Karyotypisierung sind wir nicht in der Lage, den hereditären Anophthalmus in unserer Studie einer bestimmten Art von Mutation zuzuordnen. Außerdem kann das Patau-Syndrom, das eine der wichtigsten Ätiologien ist, bei unseren Patienten klinisch nicht nachgewiesen werden.
Von all diesen ätiologischen Faktoren scheint die Theorie der genetischen Mutation zu unseren klinischen Fällen zu passen, denn diese drei Fälle stammen aus derselben Familie, aber auch aus einer konsanguinen Verbindung, da sie selbst aus derselben Familie stammen, da sie Cousins sind. Es sei jedoch darauf hingewiesen, dass Vitamin-A-Mangel, Röntgenstrahlung, missbräuchliche Verwendung von Lösungsmitteln, die Einnahme von Thalidomin und während der Schwangerschaft erworbene Infektionen ebenfalls Umweltfaktoren sein könnten. Die Diagnose einer Anophthalmie kann prä- oder postnatal auf der Grundlage einer Kombination aus klinischen Anzeichen, bildgebenden Verfahren und genetischer Analyse gestellt werden. Dufit hat unter Verwendung neuer bildgebender Verfahren (fetale MRT) einen Fall von fetalem Anophthalmus in Verbindung mit multiplen Fehlbildungen beschrieben, der zum Abbruch der Schwangerschaft führte.
Bei Kindern wird eine prothetische Behandlung durch Vergrößerung der Augenhöhle oder sogar eine Erweiterung der Augenhöhle durch aufblasbare Prothesen vor dem Alter von 2 Jahren empfohlen. Bei Erwachsenen ist diese Art der Behandlung jedoch unnötig und es wird davon abgeraten. Das Therapieschema muss eine Vergrößerung der knöchernen Augenhöhle durch verschiedene Osteotomien umfassen, die gleichzeitig oder sekundär mit der Wiederherstellung des Bindehautsacks durch Schleimhauttransplantate oder dermo-epidermale Transplantate verbunden sind. Anschließend erfolgt die Wiederherstellung der Augenlider durch Rekonstruktion, Neupositionierung und Korrektur der Fehlstellung auf der Prothese. In unseren Entwicklungsländern ist diese Art der schweren und manchmal enttäuschenden Behandlung von manchmal ungeschulten Teams nur schwer zu bewerkstelligen. Außerdem schränkt die Armut der Eltern den Zugang zur Behandlung ein, was ästhetische Folgen und erhebliche soziale Auswirkungen hat.
Schlussfolgerung
Anophthalmie ist extrem selten. Ihre Diagnose erfordert die Untersuchung sowohl von assoziierten Ätiologien als auch von Fehlbildungen. Die Behandlung ist schwer und manchmal für Kinder aus ungünstigen sozialen Verhältnissen nicht zugänglich, was zu ästhetischen Vorurteilen führt. Eine qualifizierte Ausbildung für die Pränataldiagnose, eine frühzeitige Behandlung durch geschulte Fachleute und eine angemessene technische Plattform sind die wesentlichen Mittel, um den sozialen Schaden zu verringern.
Ethikerklärung
Dieser Fallbericht folgte den Grundsätzen der Deklaration von Helsinki und wurde vom Zentralkrankenhaus der Universität Bouaké genehmigt. Die Zustimmung der Eltern des Neugeborenen wurde eingeholt und es besteht kein Interessenkonflikt bezüglich dieses Artikels.
- Speeg-Schatz, Le Marec B. Erreurs génétiques et œil in: Flamment J, Storck D. Œil et pathologie générale Rapport de la soc Fr Ophtalmol. Masson ed, Paris; 1997. P27
- Guthoff R, Klein R, Lieb WE (2004) Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 242: 268-271.
- Mouriaux F, Audo I, Defoort-Dhellemmes S, Labalette P, Guilbert F, et al. (1997) . J Fr Ophtalmol 20: 583-591.
- Roth P, Roth A, Riethmuller D. Ophtalmologie fœtale: l’utilité de l’examen des yeux au cours de l’échographie anténatale. Ophtalmologie 1997; 11: 85-96.
- Romero Caballero MD, López Soler JA, Alcázar Cantos A (2002) . Arch Soc Esp Oftalmol 77: 571-574.
- Busby A, Dolk H, Collin R, Jones RB, Winter R (1998) Compiling a national register of babies born with anophthalmia/microphthalmia in England 1988-94. Arch Dis Child Fetal Neonatal Ed 79: F168-173.
- Araujo J E, Kawanami TE, Nardozza LM, Milani HJ. Oliveira PS, Monron AF (2012) Pränatale Diagnose von bilateralem Anophthalmus durch 3D „reverse face“ Ansicht Ultraschall und Magnetresonanztomographie. Taiwan J Obstet Gynecol 51: 616-619.
- Verma AS, Fitzpatrick DR (2007) Anophthalmie und Mikrophthalmie. Orphanet J Rare Dis 2: 47.
- Kouassi FX, Koffi KV, Safede K, Cochard C, Cochener B (2006) . J Fr Ophtalmol 29: e10.
- Diomande I.A, Toure.A, Koffi. K.V, Diomande.G F, Djiguimde.W P, N Habib, A. Ahnoux-zabsonre (2015) Anophthalmia and serious microphthalmia: a summary of the problems associated with antenatal diagnosis and therapeutic refunding in Sub-Saharan. Africa International Medical Case Reports Journal 8: 287-290.
- Stoll C, Dott B, Alembik Y, Roth MP (2012) Associated malformations among infants with anophthalmia and microphthalmia. Birth Defects Res A Clin Mol Teratol 94: 147-152.12.
- Stricker M, Gola R (1990) L’anophtalmie congénitale Chirurgie plastique et réparatrice des paupières et de leurs annexes.Paris : Masson, 177-191.
- Morax.S (1998) Anophtalmies microphtalmies in: Adenis JP, Morax S. Pathologie orbito-palpébrale. Rapport de la Soc Fr Ophtalmol. Paris: Masson. 635-645.
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, et al. (2003) Mutationen in SOX2 verursachen Anophthalmie. Nat Genet 33: 461-463.
- Lee B, Song H, Rizzoti K, Son Y, Yoon J, et al. (2013) Genomic code for Sox2 binding uncovers its regulatory role in Six3 activation in the forebrain. Dev Biol 381: 491-501.
- Ye FX, Fan XQ (2012) . Zhonghua Yan Ke Za Zhi 48: 1049-1052.
- Schilter KF, Reis LM, Schneider A, Bardakjian TM, Abdul-Rahman O, et al. (2013) Whole-genome copy number variation analysis in anophthalmia and microphthalmia. Clin Genet 84: 473-481.
- You T, Lv Y, Liu S, Li F, Zhao Y, et al. (2012) Novel OTX2 mutation associated with congenital anophthalmia and microphthalmia in a Han Chinese family. Acta Ophthalmol 90: e501-502.
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, et al. (2004) Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum Mol Genet 13: 315-322.
- Gallardo ME, Rodríguez De Córdoba S, Schneider AS, Dwyer MA, Ayuso C, et al. (2004) Analysis of the developmental SIX6 homeobox gene in patients with anophthalmia/microphthalmia. Am J Med Genet A 129A: 92-94.
- Dufit C, Baggio E, Ruban JM, Buenerd A, Hermier M (1999) Fetal anophthalmos. Ein Problem der pränatalen Diagnose: ein Fallbericht. J Fr Ophtalmol 22: 966-969.