Abstract
Introduzione: L’anoftalmia congenita è l’assenza clinica dell’occhio alla nascita. Risulta dalla mancanza di sviluppo o dalla regressione della vescicola ottica primaria durante la vita embrionale. È raro e può essere isolato o associato ad altri difetti di nascita oculari o generali.
Osservazioni: Segnaliamo i casi di tre bambini della stessa famiglia di età compresa tra 4 mesi e 8 anni con anoftalmia congenita bilaterale. La loro storia è caratterizzata da una mancanza di consultazione e test di laboratorio prenatale. L’esame oftalmologico ha oggettivato, aprendo le palpebre con due occhi, un riempimento della cavità da parte del tessuto congiuntivale in 3 casi suggestivi di anoftalmia bilaterale, confermato l’ecografia oculare.
Discussione: L’anoftalmia congenita è una malformazione rara. Può essere isolata o integrata a una sindrome malformativa. Il quadro clinico è per lo più unilaterale. La diagnosi è principalmente clinica e confermata dall’ecografia oculare. Le cause sono varie, rappresentate da aberrazioni cromosomiche, mutazioni genetiche, avvelenamento e infezioni acquisite durante la gravidanza.
Conclusione: La scoperta di un’anoftalmia congenita impone una revisione completa al fine di cercare l’eziologia. Anche il sostegno psicologico dei genitori rimane un obiettivo importante.
Parole chiave
anoftalmia, congenita, ecografia oculare, aberrazioni cromosomiche
Introduzione
L’anoftalmia è la mancanza clinica dell’occhio. È un’anomalia oculare estremamente rara. Ci sono numerose eziologie, come aberrazioni cromosomiche, mutazioni genetiche, intossicazioni e infezioni. L’anoftalmia congenita può essere isolata o associata a gravi sindromi polimorformative. È responsabile, da un lato, di una micro-orbita e di un’emiatrofia cranio-facciale omolaterale e, dall’altro, di uno scarso sviluppo delle appendici che pone problemi durante l’adattamento protesico. La diagnosi è stata migliorata dall’imaging prenatale e la gestione terapeutica prevede essenzialmente protesi orbitali. Segnaliamo 3 casi di anoftalmia congenita bilaterale nella stessa famiglia.
Caso 1
Si tratta di un bambino maschio di 8 anni, nato a termine, ricevuto in consultazione per una diminuzione bilaterale dell’acuità visiva. La valutazione prenatale biologica e la TAC orbito-cerebrale non erano possibili. L’esame clinico oftalmologico ha rivelato un’assenza di percezione luminosa in entrambi gli occhi; anoftalmia bilaterale; le appendici sono presenti e normali e all’apertura delle palpebre, una struttura congiuntivale che riempie l’intera cavità orbitale senza bulbo oculare. La TAC orbito-cerebrale richiesta ha confermato l’assenza del globo oculare con presenza di grasso residuo, muscoli oculomotori e un’atrofia del calibro del nervo ottico nella cavità orbitale (Figura 1). Il bilancio complessivo era normale. E la risonanza magnetica non poteva essere realizzata considerando le difficoltà economiche dei genitori, così come il bilancio genetico (cariotipizzazione). Il trattamento chirurgico con posizionamento di una protesi è stato proposto ma non si è concretizzato a causa della mancanza di una piattaforma tecnica adeguata. (Figura 2)
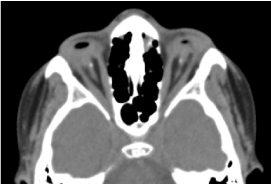
Figura.1. TC cerebrale orbitale: assenza del globo oculare con residui di grasso, muscoli e un’atrofia del calibro del nervo otocervicale nella cavità orbitale.

Figura. 2. Bambino di 8 anni che mostra anoftalmo bilaterale all’apertura delle palpebre
Caso 2
Si trattava di un bambino maschio di 8 anni, nel quale non era stata effettuata alcuna valutazione prenatale (biologica ed ecografica). È stato ricevuto in consultazione oftalmologica per una diminuzione bilaterale dell’acuità visiva. L’esame clinico oftalmologico ha trovato un’assenza di percezione luminosa in entrambi gli occhi; un’anoftalmia bilaterale, appendici senza particolarità e all’apertura delle palpebre, una struttura congiuntivale che riempie la cavità). La TAC orbito-cerebrale richiesta ha confermato l’assenza del globo oculare con la presenza di grasso residuo, muscoli oculomotori e un’atrofia del calibro del nervo ottico nella cavità orbitale (Figura 3). L’esame generale di questo bambino era normale. E la risonanza magnetica così come il cariotipo non potevano essere eseguiti per le stesse ragioni menzionate in precedenza, così come la gestione medica. (Figura 4)
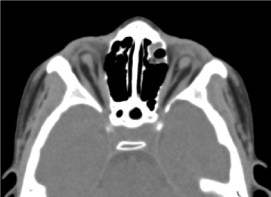
Figura.3Assenza del globo oculare con residui di grasso, muscoli e atrofia del calibro del nervo otocervicale nella cavità orbitale.
Caso 3
È quello di una neonata di 4 mesi, nata a termine e con un buon sviluppo psicomotorio e peso staturo, ricevuta in consultazione oftalmologica per una diminuzione bilaterale dell’acuità visiva e occlusione permanente delle palpebre dalla nascita. L’anamnesi del neonato è segnata da due consultazioni prenatali, ma nessun esame biologico ed ecografico prenatale. L’esame clinico oftalmologico ha trovato un’assenza di tracciamento luminoso; anoftalmia bilaterale; aspetti normali delle appendici e dell’apertura delle palpebre, un globo oculare vuoto che dà un aspetto microftalmico, i dettagli dell’iride non sono analizzabili e un’assenza di camera anteriore. La consultazione pediatrica alla ricerca di altre malformazioni è tornata senza particolarità sul piano clinico.
La tomografia orbito-cerebrale richiesta ha confermato l’assenza di un globo oculare con la presenza di grasso residuo, muscoli oculomotori e un’atrofia del calibro del nervo ottico nella cavità orbitale (Figura 5). La risonanza magnetica e la cariotipizzazione non hanno potuto essere eseguite; così come la sua gestione medica (Figura 6).

Figura.4. Bambino di 6 anni con anoftalmia bilaterale all’apertura delle palpebre.
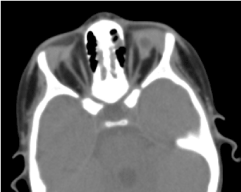
Figura. 5. Un’assenza di globo oculare con una vescicola residua, muscoli e un’atrofia del nervo otocervicale nella cavità orbitale.

Figura.6. bambino di 4 mesi; all’apertura delle palpebre si poteva notare, soprattutto nell’occhio destro un abbozzo di cornea ma l’iride non è individuabile.
Discussione
L’anoftalmia congeniale o assenza clinica dell’occhio alla nascita è una malformazione rara. La sua incidenza è variabile. È stimata a meno di un caso su 10000 nascite per Mouriaux e Roth . Sarebbe circa 21,34 per 100.000 nascite secondo un’indagine spagnola e 10 per 100.000 nascite secondo uno studio britannico. Per Aranjo , la sua incidenza è di 0,6 per 10000 bambini nati vivi. Questa differenza di incidenza trova la sua spiegazione nella variabilità della comparsa di questa malformazione. La frequenza dell’anoftalmia non è influenzata dalla razza o dal sesso. L’anoftalmia può essere isolata o integrata in una sindrome polimalformativa in un terzo dei casi. Questa osservazione è conforme a quelle di diversi autori. Così, per Verna, sia l’anoftalmia che la microftalmia possono presentarsi isolatamente o essere integrate in una sindrome di Patau (trisomia13) o in altre sindromi in circa un terzo dei casi. Kouassi ha anche riportato un caso di anoftalmia congenita bilaterale durante la sindrome di Patau. Così fa Diomandé a Bouaké che ha riportato un caso di una vera anoftalmia congenita associata a una microftalmia grave in un neonato senza altre anomalie cliniche osservate.
Inoltre, Stoll ha notato che il 90% delle anoftalmie ha malformazioni associate. Pertanto, in presenza di qualsiasi anoftalmia congenita o microftalmia estrema, è importante eseguire una valutazione pediatrica approfondita alla ricerca di anomalie associate, a volte gravi come l’anencefalia o l’agenesia dell’intero tratto ottico. L’anoftalmia clinica è raramente bilaterale. Le moderne indagini paracliniche come la tomografia computerizzata e la risonanza magnetica permettono di studiare le malformazioni associate. Nelle nostre osservazioni, questi equilibri non hanno potuto essere realizzati date le difficoltà socio-economiche dei genitori.
Secondo Speeg – Schatz , ci sono tre tipi di anoftalmia congenita: anoftalmia primaria, anoftalmo secondario e anoftalmia secondaria consecutiva come risultato di assenza di invaginazione della fossa ottica, la cessazione completa dello sviluppo del tubo neurale anteriore o la degenerazione della vescicola ottica dopo la sua invaginazione. Per Stricker , l’anoftalmia congenita potrebbe essere analizzata e classificata secondo due modalità.
Dal punto di vista anatomico, sarebbe necessario distinguere la vera anoftalmia dalle embriopatie caratterizzate dall’assenza di qualsiasi contorno oculare. Solo l’esame istologico lo confermerebbe.
Dal punto di vista clinico, l’anoftalmia clinica o microftalmia estrema deve essere separata da altre microftalmie più moderate con prognosi funzionali talvolta migliori. In questi ultimi casi, c’è un bulbo oculare più o meno rudimentale (come nel nostro 3° caso clinico) talvolta accompagnato da una cisti colobomatosa capace di fungere da protesi espansiva fisiologica. Clinicamente, non c’è struttura oculare, ma un’ecografia oculare, una tomografia computerizzata e una risonanza magnetica mostrerebbero la presenza di un abbozzo del bulbo oculare e del nervo ottico.
Tuttavia, Morax , notava che la vera anoftalmia non esiste. Secondo lui, nei casi clinici più marcati, la neuroimmagine mostrerebbe la presenza di un nervo ottico e di un minuscolo abbozzo oculare. Nei nostri primi due casi clinici, l’ecografia oculare ha mostrato un’assenza di abbozzo oculare. Tuttavia, in assenza dei risultati della TAC e della risonanza magnetica, non possiamo concludere formalmente che non c’è un nervo ottico oculare e non c’è un nervo ottico.L’eziologia dell’anoftalmia è complessa e diversa. Può essere ereditaria per aberrazioni cromosomiche, in particolare la trisomia 13 o la trisomia 18. In questo caso, si tratta di anoftalmia con malformazioni cerebrali o associata a una displasia della linea mediana. Altre eziologie genetiche sono rappresentate da mutazioni cromosomiche che possono verificarsi durante le delezioni cromosomiche o indipendentemente da qualsiasi aberrazione del cariotipo. Le anomalie SOX2 sono le principali. Fantes ha diagnosticato una microdelezione associata ad una traslocazione t (3; 11) (q26-3 p11-2) e mutazione del gene SOX2 in un paziente con anoftalmia bilaterale. L’aploinsufficienza di SOX2 è ritenuta responsabile di anoftalmia, microftalmia, displasia setto-ottica e un difetto nello sviluppo del cervello anteriore. Ye ha notato che il difetto di SOX2 causerebbe un disordine multiplo del sistema organico tra cui anoftalmia e microftalmia. Altri geni, come PAX6, OTX2, CHV10, RAX2, SIX6, NF1, TWIST, SHH, BMP4 e BMP7, sarebbero coinvolti nella comparsa dell’anoftalmia. Secondo Schilter, meno del 40% delle mutazioni genetiche conosciute spiegherebbe l’anoftalmia. Per lui, una mutazione dei geni NF1, PAX6, SOX2 sarebbe responsabile dell’anoftalmia o della microftalmia. Lei ha descritto una mutazione del gene OTX2 associata ad anoftalmia e microftalmia congenita in una famiglia cinese. Veronica ha osservato, in un paziente, una mutazione del gene RAX2 responsabile dell’anoftalmia unilaterale. Le mutazioni nel gene TWIST durante le delezioni del cromosoma 7 possono portare alla disgenesi facciale con anoftalmia. L’identificazione della mutazione di un allele del gene SIX6 in un paziente con anoftalmia bilaterale è stata notata da Gallardo.
Senza cariotipizzazione, non siamo in grado di attribuire questi anoftalmi ereditari nel nostro studio ad un tipo particolare di mutazione. Inoltre, la sindrome di Patau che è una delle principali eziologie non può essere mantenuta clinicamente nei nostri pazienti.
Tra tutti questi fattori eziologici, la teoria della mutazione genetica sembra adattarsi ai nostri casi clinici, infatti, questi 3 casi provengono dalla stessa famiglia, ma anche da un accoppiamento consanguineo perché essi stessi provengono dalla stessa famiglia essendo cugini. Tuttavia, va notato che la carenza di vitamina A, l’esposizione ai raggi X, l’uso abusivo di solventi, l’assunzione di talidomin e le infezioni acquisite durante la gravidanza sarebbero anche fattori ambientali. La diagnosi di anoftalmia può essere fatta pre- o post-natale sulla base di una combinazione di segni clinici, imaging e analisi genetica. Dufit , utilizzando nuove tecniche di imaging (risonanza magnetica fetale) ha descritto un caso di anoftalmia fetale associata a malformazioni multiple che hanno portato all’interruzione della gravidanza.
Nei bambini, il trattamento protesico mediante conformatori di dimensioni crescenti, o anche tecniche di espansione orbitale mediante protesi gonfiabili, è consigliato prima dei 2 anni di età. Tuttavia, negli adulti, questo tipo di trattamento diventa inutile e sconsigliato. Il regime terapeutico deve comprendere un allargamento dell’orbita ossea mediante diverse osteotomie associate contemporaneamente o secondariamente al ripristino del sacco congiuntivale mediante innesto di mucosa o dermo-epidermico. Poi, il restauro delle palpebre avverrà con la ricostruzione, il riposizionamento e la correzione della malposizione sulla protesi. Nei nostri paesi in via di sviluppo, questo tipo di trattamento pesante e a volte deludente è difficile da realizzare da parte di squadre a volte non formate. Inoltre, il livello di povertà dei genitori limita la loro accessibilità al trattamento, con conseguenze estetiche e una grande ripercussione sociale.
Conclusione
L’anoftalmia è estremamente rara. La sua diagnosi implica l’investigazione sia delle eziologie associate che delle malformazioni. Il suo trattamento è pesante e non accessibile a volte ai bambini di classe sociale sfavorevole con conseguente pregiudizio estetico. Una formazione qualificata per la diagnosi prenatale, una gestione precoce da parte di specialisti formati e una piattaforma tecnica adeguata sono i mezzi essenziali per ridurre il danno sociale.
Dichiarazione etica
Questa relazione di casi ha seguito i principi della Dichiarazione di Helsinki ed è stata approvata dal Centro Ospedaliero Universitario di Bouaké. Il consenso è stato ottenuto dai genitori del neonato e non vi è alcun conflitto di interessi su questo articolo.
- Speeg-Schatz, Le Marec B. Erreurs génétiques et œil in: Flamment J, Storck D. Œil et pathologie générale Rapport de la soc Fr Ophtalmol. Masson ed, Paris; 1997. P27
- Guthoff R, Klein R, Lieb WE (2004) Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 242: 268-271.
- Mouriaux F, Audo I, Defoort-Dhellemmes S, Labalette P, Guilbert F, et al. (1997) . J Fr Ophtalmol 20: 583-591.
- Roth P, Roth A, Riethmuller D. Ophtalmologie fœtale: l’utilité de l’examen des yeux au cours de l’échographie anténatale. Ophtalmologie 1997; 11: 85-96.
- Romero Caballero MD, López Soler JA, Alcázar Cantos A (2002) . Arch Soc Esp Oftalmol 77: 571-574.
- Busby A, Dolk H, Collin R, Jones RB, Winter R (1998) Compilazione di un registro nazionale dei bambini nati con anoftalmia/microftalmia in Inghilterra 1988-94. Arch Dis Child Fetal Neonatal Ed 79: F168-173.
- Araujo J E, Kawanami TE, Nardozza LM, Milani HJ. Oliveira PS, Monron AF (2012) diagnosi prenatale di anoftalmia bilaterale da 3D “reverse face” vista ecografia e risonanza magnetica. Taiwan J Obstet Gynecol 51: 616-619.
- Verma AS, Fitzpatrick DR (2007) Anoftalmia e microftalmia. Orphanet J Rare Dis 2: 47.
- Kouassi FX, Koffi KV, Safede K, Cochard C, Cochener B (2006) . J Fr Ophtalmol 29: e10.
- Diomande I.A, Toure.A, Koffi. K.V, Diomande.G F, Djiguimde.W P, N Habib, A. Ahnoux-zabsonre (2015) Anoftalmia e microftalmia grave: una sintesi dei problemi associati alla diagnosi prenatale e al rimborso terapeutico in Sub-Sahariano. Africa International Medical Case Reports Journal 8: 287-290.
- Stoll C, Dott B, Alembik Y, Roth MP (2012) Malformazioni associate tra i neonati con anoftalmia e microftalmia. Birth Defects Res A Clin Mol Teratol 94: 147-152.12.
- Stricker M, Gola R (1990) L’anoftalmia congénitale Chirurgie plastique et réparatrice des paupières et de leurs annexes.Paris : Masson, 177-191.
- Morax.S (1998) Anophtalmies microphtalmies in: Adenis JP, Morax S. Pathologie orbito-palpébrale. Rapport de la Soc Fr Ophtalmol. Parigi: Masson. 635-645.
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, et al. (2003) Mutazioni in SOX2 causano anoftalmia. Nat Genet 33: 461-463.
- Lee B, Song H, Rizzoti K, Son Y, Yoon J, et al. (2013) codice genomico per il legame Sox2 scopre il suo ruolo di regolamentazione in Six3 attivazione nel proencefalo. Dev Biol 381: 491-501.
- Ye FX, Fan XQ (2012) . Zhonghua Yan Ke Za Zhi 48: 1049-1052.
- Schilter KF, Reis LM, Schneider A, Bardakjian TM, Abdul-Rahman O, et al. (2013) Whole-genome copy number variation analysis in anoftalmia e microftalmia. Clin Genet 84: 473-481.
- You T, Lv Y, Liu S, Li F, Zhao Y, et al. (2012) Novel OTX2 mutazione associata a anoftalmia congenita e microftalmia in una famiglia cinese Han. Acta Ophthalmol 90: e501-502.
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, et al. (2004) Mutazioni nel gene umano RAX homeobox in un paziente con anoftalmia e sclerocornea. Hum Mol Genet 13: 315-322.
- Gallardo ME, Rodríguez De Córdoba S, Schneider AS, Dwyer MA, Ayuso C, et al. (2004) Analisi del gene homeobox SIX6 dello sviluppo in pazienti con anoftalmia/microftalmia. Am J Med Genet A 129A: 92-94.
- Dufit C, Baggio E, Ruban JM, Buenerd A, Hermier M (1999) Anftalmo fetale. Problema della diagnosi prenatale: un rapporto di caso. J Fr Ophtalmol 22: 966-969.