Abstract
Introduction : L’anophtalmie congénitale est l’absence clinique de l’œil à la naissance. Elle résulte de l’absence de développement ou de la régression de la vésicule optique primaire au cours de la vie embryonnaire. Elle est rare et peut être isolée ou associée à d’autres anomalies congénitales oculaires ou générales.
Observations : Nous rapportons les cas de trois enfants d’une même famille âgés de 4 mois à 8 ans présentant une anophtalmie congénitale bilatérale. Leur histoire est marquée par l’absence de consultation et d’examens de laboratoire prénataux. L’examen ophtalmologique a objectivé, l’ouverture des paupières à deux yeux, un comblement de la cavité par le tissu conjonctif dans 3 cas évocateurs d’une anophtalmie bilatérale, a confirmé l’échographie oculaire.
Discussion : L’anophtalmie congénitale est une malformation rare. Elle peut être isolée ou intégrée à un syndrome malformatif. Le tableau clinique est le plus souvent unilatéral. Le diagnostic est principalement clinique et confirmé par l’échographie oculaire. Les causes sont variées, représentées par les aberrations chromosomiques, les mutations génétiques, les intoxications et les infections acquises pendant la grossesse.
Conclusion : La découverte d’une anophtalmie congénitale impose un bilan complet afin de rechercher l’étiologie. Le soutien psychologique des parents reste également un axe important.
Mots clés
anophtalmie, congénitale, échographie oculaire, aberrations chromosomiques
Introduction
L’anophtalmie est l’absence clinique de l’œil. Il s’agit d’une anomalie oculaire extrêmement rare. Les étiologies sont nombreuses, telles que les aberrations chromosomiques, les mutations génétiques, les intoxications et les infections. L’anophtalmie congénitale peut être isolée ou associée à des syndromes polymorformatifs sévères. Elle est responsable, d’une part, d’une micro-orbite et d’une hémi-atrophie cranio-faciale homolatérale et, d’autre part, d’un mauvais développement des appendices posant des problèmes lors de l’adaptation prothétique. Le diagnostic a été amélioré par l’imagerie prénatale et la prise en charge thérapeutique fait essentiellement appel aux prothèses orbitaires. Nous rapportons 3 cas d’anophtalmie congénitale bilatérale dans une même famille.
Cas 1
Il s’agit d’un enfant de sexe masculin âgé de 8 ans, né à terme, reçu en consultation pour une baisse d’acuité visuelle bilatérale. Le bilan prénatal biologique et tomodensitométrique orbite-cérébral n’a pas été possible. L’examen clinique ophtalmologique a révélé une absence de perception lumineuse aux deux yeux ; une anophtalmie bilatérale ; les appendices sont présents et normaux et à l’ouverture des paupières, une structure conjonctivale remplissant toute la cavité orbitaire sans globe oculaire. Le scanner orbito-cérébral demandé a confirmé l’absence du globe oculaire avec la présence de graisse résiduelle, de muscles oculomoteurs et une atrophie du calibre du nerf optique dans la cavité orbitaire (figure 1). Le bilan global était normal. Et l’imagerie par résonance magnétique n’a pu être réalisée compte tenu des difficultés financières des parents, ainsi que le bilan génétique (caryotype). Un traitement chirurgical avec pose d’une prothèse a été proposé mais non concrétisé faute de plateau technique adéquat. (Figure 2)
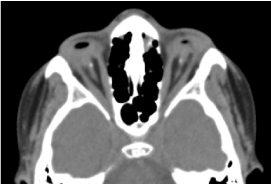
Figure.1. CT-scan cérébral orbitaire : absence du globe oculaire avec de la graisse résiduelle, des muscles et une atrophie du calibre du nerf otocervical dans la cavité orbitaire.

Figure. 2. Enfant de 8 ans présentant une anophtalmie bilatérale à l’ouverture des paupières
Cas 2
Il s’agissait d’un enfant de sexe masculin de 8 ans, chez qui aucun bilan prénatal (biologique et échographique) n’a été réalisé. Il était reçu en consultation ophtalmologique pour une baisse d’acuité visuelle bilatérale. L’examen clinique ophtalmologique a retrouvé une absence de perception lumineuse aux deux yeux ; Une anophtalmie bilatérale, des appendices sans particularités et à l’ouverture des paupières, une structure conjonctivale remplissant la cavité).Le scanner orbito-cérébral demandé a confirmé l’absence du globe oculaire avec la présence de graisse résiduelle, de muscles oculomoteurs et une atrophie du calibre du nerf optique dans la cavité orbitaire (Figure 3). L’examen général de cet enfant était normal. Et l’imagerie par résonance magnétique ainsi que le caryotype n’ont pu être réalisés pour les mêmes raisons évoquées précédemment, ainsi que la prise en charge médicale. (Figure 4)
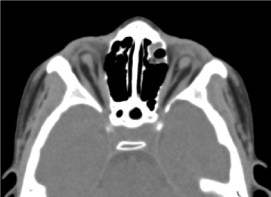
Figure.3. absence du globe oculaire avec graisse résiduelle, muscles et une atrophie du calibre du nerf otocervical dans la cavité orbitaire.
Cas 3
C’est celui d’un nourrisson de sexe féminin âgé de 4 mois, né à terme et ayant un bon développement psychomoteur et staturo-pondéral, reçu en consultation ophtalmologique pour une baisse d’acuité visuelle bilatérale et une occlusion permanente des paupières depuis la naissance. L’histoire du nourrisson est marquée par deux consultations prénatales, mais pas d’examens biologiques et échographiques prénataux. L’examen clinique ophtalmologique retrouve une absence de repérage lumineux ; une anophtalmie bilatérale ; des aspects normaux des appendices et à l’ouverture des paupières, une ébauche de globe oculaire donnant un aspect microphtalmique, les détails de l’iris ne sont pas analysables et une absence de chambre antérieure. La consultation pédiatrique à la recherche d’autres malformations est revenue sans particularité sur le plan clinique.
La tomographie orbito-cérébrale demandée a confirmé l’absence de globe oculaire avec la présence de graisse résiduelle, de muscles oculomoteurs et une atrophie du calibre du nerf optique dans la cavité orbitaire (figure 5). L’imagerie par résonance magnétique et le caryotype n’ont pu être réalisés ; ainsi que sa prise en charge médicale (Figure 6).

Figure.4. Enfant de 6 ans présentant une anophtalmie bilatérale à l’ouverture des paupières.
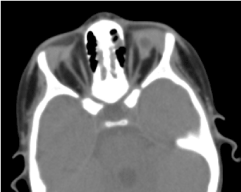
Figure. 5. Une absence de globe oculaire avec une vésicule résiduelle, des muscles et une atrophie du nerf otocervical dans la cavité orbitaire.

Figure.6.. enfant de 4 mois ; à l’ouverture des paupières on pouvait remarquer, surtout à l’œil droit une esquisse de cornée mais l’iris n’est pas individualisable.
Discussion
L’anophtalmie congénitale ou absence clinique de l’œil à la naissance est une malformation rare . Son incidence est variable. Elle est estimée à moins d’un cas pour 10000 naissances pour Mouriaux et Roth . Elle serait d’environ 21,34 pour 100 000 naissances selon une enquête espagnole et de 10 pour 100 000 naissances selon une étude britannique . Pour Aranjo , son incidence est de 0,6 pour 10000 enfants nés vivants. Cette différence d’incidence trouve son explication dans la variabilité d’apparition de cette malformation. La fréquence de l’anophtalmie n’est pas influencée par la race ou le sexe. L’anophtalmie peut être isolée ou intégrée dans un syndrome polymalformatif dans un tiers des cas. Cette observation est conforme à celles de plusieurs auteurs. Ainsi, pour Verna, l’anophtalmie et la microphtalmie peuvent être isolées ou intégrées dans un syndrome de Patau (trisomie 13) ou d’autres syndromes dans environ un tiers des cas. Kouassi rapporte également un cas d’anophtalmie congénitale bilatérale au cours du syndrome de Patau. De même que Diomandé à Bouaké qui a rapporté un cas d’une véritable anophtalmie congénitale associée à une microphtalmie sévère chez un nouveau-né sans aucune autre anomalie clinique observée.
En outre, Stoll a noté que 90% des anophtalmies avaient des malformations associées. Par conséquent, en présence de toute anophtalmie congénitale ou microphtalmie extrême, il est important d’effectuer un bilan pédiatrique complet à la recherche d’anomalies associées, parfois sévères comme une anencéphalie ou une agénésie de l’ensemble du tractus optique . L’anophtalmie clinique est rarement bilatérale. Les examens paracliniques modernes tels que la tomodensitométrie et l’imagerie par résonance magnétique permettent de rechercher les malformations associées. Dans nos observations, ces bilans n’ont pu être réalisés compte tenu des difficultés socio-économiques des parents.
Selon Speeg – Schatz , il existe trois types d’anophtalmie congénitale : l’anophtalmie primaire, l’anophtalmie secondaire et l’anophtalmie secondaire consécutive résultant de l’absence d’invagination de la fosse optique, de l’arrêt complet du développement du tube neural antérieur ou de la dégénérescence de la vésicule optique après son invagination. Pour Stricker , l’anophtalmie congénitale pourrait être analysée et classée selon deux modes.
Du point de vue anatomique, il faudrait distinguer la véritable anophtalmie des embryopathies caractérisées par l’absence de tout contour oculaire. Seul l’examen histologique permettrait de le confirmer.
Du point de vue clinique, l’anophtalmie clinique ou microphtalmie extrême doit être séparée d’autres microphtalmies plus modérées dont le pronostic fonctionnel est parfois meilleur. Dans ces derniers cas, il existe un globe oculaire plus ou moins rudimentaire (comme dans notre 3ème cas clinique) parfois accompagné d’un kyste colobomateux pouvant servir de prothèse expansive physiologique. Cliniquement, il n’y a pas de structure oculaire, mais une échographie oculaire, une tomodensitométrie et une imagerie par résonance magnétique montreraient la présence d’une ébauche du globe oculaire et du nerf optique.
Cependant, Morax , a noté que la véritable anophtalmie n’existait pas. Selon lui, dans les cas cliniques les plus marqués, la neuro-imagerie montrerait la présence d’un nerf optique et d’une minuscule esquisse oculaire. Dans nos deux premiers cas cliniques, l’échographie oculaire a montré une absence d’ébauche oculaire. Cependant, en l’absence de résultats de CT-scan et d’IRM, nous ne pouvons pas conclure formellement à l’absence de nerf optique oculaire et à l’absence de nerf optique.L’étiologie de l’anophtalmie est complexe et diverse. Elle peut être héréditaire par des aberrations chromosomiques, en particulier la trisomie 13 ou la trisomie 18. Dans ce cas, il s’agit d’une anophtalmie avec malformations cérébrales ou associée à une dysplasie de la ligne médiane . Les autres étiologies génétiques sont représentées par des mutations chromosomiques qui peuvent survenir lors de délétions chromosomiques ou indépendamment de toute aberration du caryotype. Les anomalies de SOX2 sont les principales. Fantes a diagnostiqué une microdélétion associée à une translocation t (3 ; 11) (q26-3 p11-2) et une mutation du gène SOX2 chez un patient présentant une anophtalmie bilatérale. L’haploinsuffisance de SOX2 serait responsable de l’anophtalmie, de la microphtalmie, de la dysplasie septo-optique et d’un défaut de développement du cerveau antérieur. Ye a noté que le défaut de SOX2 provoquerait un trouble multiple du système organique, notamment l’anophtalmie et la microphtalmie. D’autres gènes, tels que PAX6, OTX2, CHV10, RAX2, SIX6, NF1, TWIST, SHH, BMP4 et BMP7, seraient impliqués dans l’apparition de l’anophtalmie. Selon Schilter, moins de 40% des mutations génétiques connues expliqueraient l’anophtalmie. Pour lui, une mutation des gènes NF1, PAX6, SOX2 serait responsable d’une anophtalmie ou d’une microphtalmie. Vous avez décrit une mutation du gène OTX2 associée à une anophtalmie et une microphtalmie congénitale dans une famille chinoise. Veronica a observé, chez un patient, une mutation du gène RAX2 responsable d’une anophtalmie unilatérale. Les mutations du gène TWIST lors des délétions du chromosome 7 peuvent conduire à une dysgénésie faciale avec anophtalmie . L’identification de la mutation d’un allèle du gène SIX6 chez un patient présentant une anophtalmie bilatérale a été notée par Gallardo .
Sans caryotype, nous ne sommes pas en mesure d’attribuer ces anophtalmies héréditaires dans notre étude à un type particulier de mutation. De plus, le syndrome de Patau qui est l’une des principales étiologies ne peut être retenu cliniquement chez nos patients.
De tous ces facteurs étiologiques, la théorie de la mutation génétique semble s’adapter à nos cas cliniques, en effet, ces 3 cas sont issus d’une même famille, mais aussi d’un accouplement consanguin car eux-mêmes sont issus de la même famille car cousins d’Être. Cependant, il faut noter que la carence en vitamine A, l’exposition aux rayons X, l’utilisation abusive de solvant, la prise de thalidomine et les infections acquises pendant la grossesse seraient également des facteurs environnementaux . Le diagnostic d’anophtalmie peut être posé avant ou après la naissance sur la base d’une combinaison de signes cliniques, d’imagerie et d’analyse génétique. Dufit , en utilisant les nouvelles techniques d’imagerie (IRM fœtale) a décrit un cas d’anophtalmie fœtale associée à de multiples malformations conduisant à une interruption de grossesse.
Chez les enfants, le traitement prothétique par augmentation des conformateurs de taille, voire les techniques d’expansion orbitaire par prothèses gonflables, est recommandé avant l’âge de 2 ans. En revanche, chez l’adulte, ce type de traitement devient inutile et déconseillé. Le schéma thérapeutique doit comporter un élargissement de l’orbite osseuse par différentes ostéotomies associées dans le même temps ou secondairement à la restauration du sac conjonctival par greffe muqueuse ou dermo-épidermique. Ensuite, la restauration des paupières se fera par reconstruction, repositionnement et correction de la malposition sur la prothèse . Dans nos pays en voie de développement, ce type de traitement lourd et parfois décevant est difficile à réaliser par des équipes parfois non formées. De plus, le niveau de pauvreté des parents limite leur accessibilité au traitement, avec des conséquences esthétiques et un retentissement social important.
Conclusion
L’anophtalmie est extrêmement rare. Son diagnostic implique la recherche à la fois d’étiologies associées et de malformations. Son traitement est lourd et non accessible parfois aux enfants de classe sociale défavorable entraînant un préjudice esthétique. Une formation qualifiante pour le diagnostic prénatal, une prise en charge précoce par des spécialistes formés et un plateau technique adéquat sont les moyens essentiels pour réduire le préjudice social.
Déclaration d’éthique
Ce rapport de cas a suivi les principes de la déclaration d’Helsinki et a été approuvé par le Centre Hospitalier Universitaire de Bouaké. Le consentement a été obtenu des parents du nouveau-né et il n’y a pas de conflit d’intérêt à propos de cet article.
- Speeg-Schatz, Le Marec B. Erreurs génétiques et œil in : Flamment J, Storck D. Œil et pathologie générale Rapport de la soc Fr Ophtalmol. Masson ed, Paris ; 1997. P27
- Guthoff R, Klein R, Lieb WE (2004) Œil kystique congénital. Graefes Arch Clin Exp Ophthalmol 242 : 268-271.
- Mouriaux F, Audo I, Defoort-Dhellemmes S, Labalette P, Guilbert F, et al. (1997) . J Fr Ophtalmol 20 : 583-591.
- Roth P, Roth A, Riethmuller D. Ophtalmologie fœtale : l’utilité de l’examen des yeux au cours de l’échographie anténatale. Ophtalmologie 1997 ; 11 : 85-96.
- Romero Caballero MD, López Soler JA, Alcázar Cantos A (2002) . Arch Soc Esp Oftalmol 77 : 571-574.
- Busby A, Dolk H, Collin R, Jones RB, Winter R (1998) Compilation d’un registre national des bébés nés avec une anophtalmie/microphtalmie en Angleterre 1988-94. Arch Dis Child Fetal Neonatal Ed 79 : F168-173.
- Araujo J E, Kawanami TE, Nardozza LM, Milani HJ. Oliveira PS, Monron AF (2012) Diagnostic prénatal d’anophtalmie bilatérale par échographie 3D en vue « face inversée » et imagerie par résonance magnétique. Taiwan J Obstet Gynecol 51 : 616-619.
- Verma AS, Fitzpatrick DR (2007) Anophtalmie et microphtalmie. Orphanet J Rare Dis 2 : 47.
- Kouassi FX, Koffi KV, Safede K, Cochard C, Cochener B (2006) . J Fr Ophtalmol 29 : e10.
- Diomande I.A, Toure.A, Koffi. K.V, Diomande.G F, Djiguimde.W P, N Habib, A. Ahnoux-zabsonre (2015) Anophtalmie et microphtalmie grave : une synthèse des problèmes liés au diagnostic anténatal et au remboursement thérapeutique en Afrique subsaharienne. Africa International Medical Case Reports Journal 8 : 287-290.
- Stoll C, Dott B, Alembik Y, Roth MP (2012) Associated malformations among infants with anophthalmia and microphthalmia. Birth Defects Res A Clin Mol Teratol 94 : 147-152.12.
- Stricker M, Gola R (1990) L’anophtalmie congénitale Chirurgie plastique et réparatrice des paupières et de leurs annexes.Paris : Masson, 177-191.
- Morax.S (1998) Anophtalmies microphtalmies dans : Adenis JP, Morax S. Pathologie orbito-palpébrale. Rapport de la Soc Fr Ophtalmol. Paris : Masson. 635-645.
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, et al. (2003) Mutations in SOX2 cause anophthalmia. Nat Genet 33 : 461-463.
- Lee B, Song H, Rizzoti K, Son Y, Yoon J, et al. (2013) Le code génomique de la liaison de Sox2 dévoile son rôle régulateur dans l’activation de Six3 dans le cerveau antérieur. Dev Biol 381 : 491-501.
- Ye FX, Fan XQ (2012) . Zhonghua Yan Ke Za Zhi 48 : 1049-1052.
- Schilter KF, Reis LM, Schneider A, Bardakjian TM, Abdul-Rahman O, et al. (2013) Analyse de la variation du nombre de copies du génome entier dans l’anophtalmie et la microphtalmie. Clin Genet 84 : 473-481.
- You T, Lv Y, Liu S, Li F, Zhao Y, et al. (2012) Nouvelle mutation OTX2 associée à une anophtalmie et une microphtalmie congénitales dans une famille chinoise Han. Acta Ophthalmol 90 : e501-502.
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, et al. (2004) Mutations du gène homeobox humain RAX chez un patient atteint d’anophtalmie et de sclérocornée. Hum Mol Genet 13 : 315-322.
- Gallardo ME, Rodríguez De Córdoba S, Schneider AS, Dwyer MA, Ayuso C, et al. (2004) Analyse du gène homéobox SIX6 en développement chez des patients atteints d’anophtalmie/microphtalmie. Am J Med Genet A 129A : 92-94.
- Dufit C, Baggio E, Ruban JM, Buenerd A, Hermier M (1999) Anophtalmie fœtale. Problème du diagnostic prénatal : un rapport de cas. J Fr Ophtalmol 22 : 966-969.