Abstract
Introducción: La anoftalmia congénita es la ausencia clínica del ojo al nacer. Es el resultado de la falta de desarrollo o regresión de la vesícula óptica primaria durante la vida embrionaria. Es rara y puede estar aislada o asociada a otros defectos oculares o generales de nacimiento.
Observaciones: Comunicamos los casos de tres niños de una misma familia con edades comprendidas entre los 4 meses y los 8 años con anoftalmia congénita bilateral. Su historia está marcada por la falta de consulta y pruebas de laboratorio prenatales. El examen oftalmológico objetivó, abriendo los párpados con los dos ojos, un relleno de la cavidad por el tejido conjuntival en 3 casos sugestivo de anoftalmia bilateral, confirmado la ecografía ocular.
Discusión: La anoftalmia congénita es una malformación poco frecuente. Puede ser aislada o integrada a un síndrome de malformación. El cuadro clínico es mayoritariamente unilateral. El diagnóstico es principalmente clínico y se confirma con una ecografía ocular. Las causas son variadas, representadas por aberraciones cromosómicas, mutaciones genéticas, intoxicaciones e infecciones adquiridas durante el embarazo.
Conclusión: El descubrimiento de una anoftalmia congénita impone una revisión exhaustiva para buscar la etiología. El apoyo psicológico de los padres también sigue siendo un foco importante.
Palabras clave
anoftalmia, congénita, ecografía ocular, aberraciones cromosómicas
Introducción
La anoftalmia es la ausencia clínica del ojo. Es una anomalía ocular extremadamente rara. Existen numerosas etiologías, como aberraciones cromosómicas, mutaciones genéticas, intoxicaciones e infecciones. La anoftalmia congénita puede ser aislada o estar asociada a síndromes polimorfos graves. Es responsable, por un lado, de una microórbita y de una hemiatrofia craneofacial homolateral y, por otro, de un mal desarrollo de los apéndices que plantea problemas durante la adaptación protésica. El diagnóstico ha mejorado gracias a las imágenes prenatales y el tratamiento terapéutico pasa esencialmente por las prótesis orbitarias. Comunicamos 3 casos de anoftalmia congénita bilateral en una misma familia.
Caso 1
Se trata de un niño varón de 8 años, nacido a término, recibido en consulta por una disminución bilateral de la agudeza visual. No fue posible la valoración prenatal biológica y por TAC órbito-cerebral. El examen clínico oftalmológico reveló ausencia de percepción luminosa en ambos ojos; anoftalmia bilateral; los apéndices están presentes y son normales y en la apertura de los párpados, una estructura conjuntival que llena toda la cavidad orbitaria sin globo ocular. El TAC órbito-cerebral solicitado ha confirmado la ausencia del globo ocular con la presencia de grasa residual, músculos oculomotores y una atrofia del calibre del nervio óptico en la cavidad orbitaria (Figura 1). El balance general era normal. Y la resonancia magnética no pudo realizarse teniendo en cuenta las dificultades económicas de los padres, así como el balance genético (cariotipo). Se propuso un tratamiento quirúrgico con colocación de una prótesis que no se materializó por falta de plataforma técnica adecuada. (Figura 2)
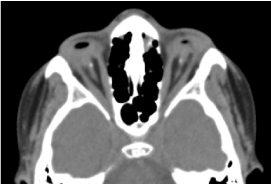
Figura.1. TAC cerebral de la órbita: ausencia del globo ocular con grasa residual, músculos y una atrofia del calibre del nervio otocerval en la cavidad orbitaria.

Figura. 2. Niño de 8 años que presenta anoftalmos bilateral en la apertura de los párpados
Caso 2
Se trata de un niño varón de 8 años, en el que no se realizó valoración prenatal (biológica y ecográfica). Fue recibido en consulta oftalmológica por una disminución bilateral de la agudeza visual. En el examen clínico oftalmológico se encontró una ausencia de percepción luminosa en ambos ojos; una anoftalmia bilateral, apéndices sin particularidades y en la apertura de los párpados, una estructura conjuntival que rellenaba la cavidad).La TC órbito-cerebral solicitada ha confirmado la ausencia del globo ocular con la presencia de grasa residual, músculos oculomotores y una atrofia del calibre del nervio óptico en la cavidad orbitaria (Figura 3). La exploración general de este niño era normal. Y la resonancia magnética así como el cariotipo no se pudieron realizar por las mismas razones mencionadas anteriormente, así como el manejo médico. (Figura 4)
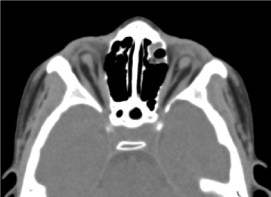
Figura.3Ausencia del globo ocular con grasa residual, músculos y atrofia del calibre del nervio otocerval en la cavidad orbitaria.
Caso 3
Es el de una lactante de 4 meses de edad, nacida a término y con buen desarrollo psicomotor y estaturo-peso, recibida en consulta oftalmológica por disminución bilateral de la agudeza visual y oclusión permanente de los párpados desde el nacimiento. Los antecedentes del bebé están marcados por dos consultas prenatales, pero ninguna exploración biológica y ecográfica prenatal. El examen clínico oftalmológico encontró una ausencia de rastreo luminoso; anoftalmia bilateral; aspectos normales de los apéndices y a la apertura de los párpados, un globo ocular en blanco dando un aspecto microftálmico, los detalles del iris no son analizables y una ausencia de cámara anterior. La consulta pediátrica en busca de otras malformaciones volvió sin ninguna particularidad a nivel clínico.
La tomografía órbito-cerebral solicitada ha confirmado la ausencia de globo ocular con la presencia de grasa residual, músculos oculomotores y una atrofia del calibre del nervio óptico en la cavidad orbitaria (Figura 5). La resonancia magnética y el cariotipo no pudieron realizarse; así como su manejo médico (Figura 6).

Figura.4. Niño de 6 años con anoftalmia bilateral en la apertura de los párpados.
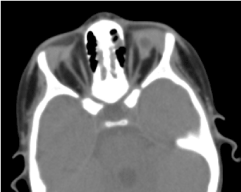 Figura. 5. Ausencia de globo ocular con una vesícula residual, músculos y una atrofia del nervio otocerval en la cavidad orbitaria.
Figura. 5. Ausencia de globo ocular con una vesícula residual, músculos y una atrofia del nervio otocerval en la cavidad orbitaria.

Figura.6. niño de 4 meses; al abrir los párpados se nota, sobre todo en el ojo derecho un esbozo de córnea pero el iris no es individualizable.
Discusión
La anoftalmia congénita o ausencia clínica del ojo al nacer es una malformación rara . Su incidencia es variable. Se estima en menos de un caso por cada 10000 nacimientos para Mouriaux y Roth . Sería de unos 21,34 por 100.000 nacimientos según una encuesta española y de 10 por 100.000 nacimientos según un estudio británico . Para Aranjo , su incidencia es de 0,6 por cada 10000 niños nacidos vivos. Esta diferencia de incidencia encuentra su explicación en la variabilidad de aparición de esta malformación. La frecuencia de la anoftalmia no está influenciada por la raza o el sexo. La anoftalmia puede estar aislada o integrada en un síndrome polimalformativo en un tercio de los casos. Esta observación está en conformidad con las de varios autores. Así, en el caso de Verna, tanto la anoftalmia como la microftalmia pueden presentarse de forma aislada o integrarse en un síndrome de Patau (trisomía13) o en otros síndromes en aproximadamente un tercio de los casos. Kouassi también informó de un caso de anoftalmia congénita bilateral durante el síndrome de Patau. También lo hace Diomandé en Bouaké, quien informó de un caso de una verdadera anoftalmia congénita asociada a una microftalmia grave en un recién nacido sin ninguna otra anomalía clínica observada.
Además, Stoll señaló que el 90% de las anoftalmias tenían malformaciones asociadas. Por lo tanto, en presencia de cualquier anoftalmia congénita o microftalmia extrema, es importante realizar una evaluación pediátrica exhaustiva en busca de anomalías asociadas, a veces graves como la anencefalia o la agenesia de todo el tracto óptico . La anoftalmia clínica rara vez es bilateral. Las investigaciones paraclínicas modernas, como la tomografía computarizada y la resonancia magnética, permiten investigar las malformaciones asociadas. En nuestras observaciones, estos balances no pudieron realizarse dadas las dificultades socioeconómicas de los padres.
Según Speeg – Schatz , existen tres tipos de anoftalmia congénita: la anoftalmia primaria, el anoftalmos secundario y la anoftalmia secundaria consecutiva como resultado de la ausencia de invaginación de la fosa óptica, el cese completo del desarrollo del tubo neural anterior o la degeneración de la vesícula óptica después de su invaginación. Para Stricker , la anoftalmia congénita podría analizarse y clasificarse según dos modalidades.
Desde el punto de vista anatómico, habría que distinguir la verdadera anoftalmia de las embriopatías caracterizadas por la ausencia de todo contorno ocular. Sólo el examen histológico lo confirmaría.
Desde el punto de vista clínico, hay que separar la anoftalmia clínica o microftalmia extrema de otras microftalmias más moderadas con un pronóstico funcional a veces mejor. En estos últimos casos, existe un globo ocular más o menos rudimentario (como en nuestro 3er caso clínico) a veces acompañado de un quiste colobomatoso capaz de actuar como prótesis expansiva fisiológica. Clínicamente, no hay estructura ocular, pero una ecografía ocular, una tomografía computarizada y una resonancia magnética mostrarían la presencia de un esbozo del globo ocular y del nervio óptico.
Sin embargo, Morax , señaló que no existía una verdadera anoftalmia. Según él, en los casos clínicos más marcados, la neuroimagen mostraría la presencia de un nervio óptico y un diminuto esbozo ocular. En nuestros dos primeros casos clínicos, la ecografía ocular mostró una ausencia de esbozo ocular. Sin embargo, en ausencia de los resultados de la tomografía y la resonancia magnética, no podemos concluir formalmente que no hay nervio óptico ocular ni nervio óptico.La etiología de la anoftalmia es compleja y diversa. Puede ser hereditaria por aberraciones cromosómicas, en particular la trisomía 13 o la trisomía 18. En este caso, se trata de una anoftalmia con malformaciones cerebrales o asociada a una displasia de la línea media . Otras etiologías genéticas están representadas por mutaciones cromosómicas que pueden producirse durante las deleciones cromosómicas o independientemente de cualquier aberración del cariotipo. Las anomalías SOX2 son las principales. Fantes diagnosticó una microdeleción asociada a una translocación t (3; 11) (q26-3 p11-2) y una mutación del gen SOX2 en una paciente con anoftalmia bilateral. Se cree que la haploinsuficiencia de SOX2 es responsable de la anoftalmia, la microftalmia, la displasia septo-óptica y un defecto en el desarrollo del cerebro anterior . Ye señaló que el defecto de SOX2 causaría un trastorno múltiple del sistema orgánico que incluye anoftalmia y microftalmia. Otros genes, como PAX6, OTX2, CHV10, RAX2, SIX6, NF1, TWIST, SHH, BMP4 y BMP7, estarían implicados en la aparición de la anoftalmia. Según Schilter , menos del 40% de las mutaciones genéticas conocidas explicarían la anoftalmia. Para él, una mutación de los genes NF1, PAX6, SOX2 sería responsable de la anoftalmia o microftalmia. Describió una mutación del gen OTX2 asociada a la anoftalmia y a la microftalmia congénita en una familia china. Verónica observó, en un paciente, una mutación del gen RAX2 responsable de una anoftalmia unilateral. Las mutaciones en el gen TWIST durante las deleciones del cromosoma 7 pueden provocar disgenesia facial con anoftalmia . La identificación de la mutación de un alelo del gen SIX6 en un paciente con anoftalmia bilateral fue señalada por Gallardo .
Sin cariotipo, no podemos atribuir estos anoftalmos hereditarios en nuestro estudio a un tipo de mutación particular. Además, el síndrome de Patau que es una de las principales etiologías no puede ser retenido clínicamente en nuestros pacientes.
De todos estos factores etiológicos, la teoría de la mutación genética parece encajar con nuestros casos clínicos, en efecto, estos 3 casos provienen de la misma familia, pero también de un acoplamiento consanguíneo porque ellos mismos provienen de la misma familia al ser primos. Sin embargo, hay que tener en cuenta que la carencia de vitamina A, la exposición a los rayos X, el uso abusivo de disolventes, la ingesta de talidominas y las infecciones adquiridas durante el embarazo serían también factores ambientales . El diagnóstico de la anoftalmia puede hacerse antes o después del nacimiento, basándose en una combinación de signos clínicos, imágenes y análisis genéticos. Dufit , utilizando nuevas técnicas de imagen (resonancia magnética fetal) ha descrito un caso de anoftalmia fetal asociada a múltiples malformaciones que condujo a la interrupción del embarazo.
En los niños, el tratamiento protésico mediante conformadores de aumento de tamaño, o incluso técnicas de expansión orbitaria mediante prótesis hinchables, se recomienda antes de los 2 años de edad. Sin embargo, en los adultos, este tipo de tratamiento resulta innecesario y se desaconseja. El régimen terapéutico debe incluir una ampliación de la órbita ósea mediante diferentes osteotomías asociadas al mismo tiempo o secundariamente a la restauración del saco conjuntival mediante injerto de mucosa o injerto dermoepidérmico. A continuación, se procederá a la restauración de los párpados mediante la reconstrucción, el reposicionamiento y la corrección de la malposición en la prótesis . En nuestros países en vías de desarrollo, este tipo de tratamiento pesado y a veces decepcionante es difícil de realizar por equipos a veces no formados. Además, el nivel de pobreza de los padres limita su accesibilidad al tratamiento, con consecuencias estéticas y una repercusión social importante.
Conclusión
La anoftalmia es extremadamente rara. Su diagnóstico implica la investigación tanto de etiologías como de malformaciones asociadas. Su tratamiento es pesado y no accesible a veces a los niños de clase social desfavorable lo que da lugar a prejuicios estéticos. La formación cualificada para el diagnóstico prenatal, el tratamiento precoz por especialistas formados y una plataforma técnica adecuada son los medios esenciales para reducir el perjuicio social.
Declaración ética
Este informe de casos siguió los principios de la Declaración de Helsinki y fue aprobado por el Centro Hospitalario Universitario de Bouaké. Se obtuvo el consentimiento de los padres del recién nacido y no hay ningún conflicto de intereses sobre este artículo.
- Speeg-Schatz, Le Marec B. Erreurs génétiques et œil in: Flamment J, Storck D. Œil et pathologie générale Rapport de la soc Fr Ophtalmol. Masson ed, París; 1997. P27
- Guthoff R, Klein R, Lieb WE (2004) Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 242: 268-271.
- Mouriaux F, Audo I, Defoort-Dhellemmes S, Labalette P, Guilbert F, et al. (1997) . J Fr Ophtalmol 20: 583-591.
- Roth P, Roth A, Riethmuller D. Ophtalmologie fœtale: l’utilité de l’examen des yeux au cours de l’échographie anténatale. Ophtalmologie 1997; 11: 85-96.
- Romero Caballero MD, López Soler JA, Alcázar Cantos A (2002) . Arch Soc Esp Oftalmol 77: 571-574.
- Busby A, Dolk H, Collin R, Jones RB, Winter R (1998) Compiling a national register of babies born with anophthalmia/microphthalmia in England 1988-94. Arch Dis Child Fetal Neonatal Ed 79: F168-173.
- Araujo J E, Kawanami TE, Nardozza LM, Milani HJ. Oliveira PS, Monron AF (2012) Diagnóstico prenatal de anoftalmia bilateral por ultrasonido de vista de «cara invertida» en 3D y resonancia magnética. Taiwan J Obstet Gynecol 51: 616-619.
- Verma AS, Fitzpatrick DR (2007) Anophthalmia and microphthalmia. Orphanet J Rare Dis 2: 47.
- Kouassi FX, Koffi KV, Safede K, Cochard C, Cochener B (2006) . J Fr Ophtalmol 29: e10.
- Diomande I.A, Toure.A, Koffi. K.V, Diomande.G F, Djiguimde.W P, N Habib, A. Ahnoux-zabsonre (2015) Anoftalmia y microftalmia grave: un resumen de los problemas asociados con el diagnóstico prenatal y el reembolso terapéutico en el subsahariano. Africa International Medical Case Reports Journal 8: 287-290.
- Stoll C, Dott B, Alembik Y, Roth MP (2012) Associated malformations among infants with anophthalmia and microphthalmia. Birth Defects Res A Clin Mol Teratol 94: 147-152.12.
- Stricker M, Gola R (1990) L’anophtalmie congénitale Chirurgie plastique et réparatrice des paupières et de leurs annexes.Paris : Masson, 177-191.
- Morax.S (1998) Anophtalmies microphtalmies in: Adenis JP, Morax S. Pathologie orbito-palpébrale. Rapport de la Soc Fr Ophtalmol. Paris: Masson. 635-645.
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, et al. (2003) Mutations in SOX2 cause anophthalmia. Nat Genet 33: 461-463.
- Lee B, Song H, Rizzoti K, Son Y, Yoon J, et al. (2013) El código genómico para la unión de Sox2 descubre su papel regulador en la activación de Six3 en el cerebro anterior. Dev Biol 381: 491-501.
- Ye FX, Fan XQ (2012) . Zhonghua Yan Ke Za Zhi 48: 1049-1052.
- Schilter KF, Reis LM, Schneider A, Bardakjian TM, Abdul-Rahman O, et al. (2013) Whole-genome copy number variation analysis in anophthalmia and microphthalmia. Clin Genet 84: 473-481.
- You T, Lv Y, Liu S, Li F, Zhao Y, et al. (2012) Nueva mutación de OTX2 asociada a la anoftalmia congénita y microftalmia en una familia china Han. Acta Ophthalmol 90: e501-502.
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, et al. (2004) Mutaciones en el gen homeobox RAX humano en un paciente con anoftalmia y esclerocórnea. Hum Mol Genet 13: 315-322.
- Gallardo ME, Rodríguez De Córdoba S, Schneider AS, Dwyer MA, Ayuso C, et al. (2004) Análisis del gen homeobox SIX6 del desarrollo en pacientes con anoftalmia/microftalmia. Am J Med Genet A 129A: 92-94.
- Dufit C, Baggio E, Ruban JM, Buenerd A, Hermier M (1999) Fetal anophthalmos. Problema del diagnóstico prenatal: informe de un caso. J Fr Ophtalmol 22: 966-969.