In der routinemäßigen Laborpraxis wird die Diagnose Beta-Thalassämie-Typ in der Regel durch charakteristische Befunde in der Hämoglobinauswertung und im Erythrozytenzahl- und -indizes gestellt. Insbesondere ist der prozentuale Anteil von Hämoglobin (Hb) A2 erhöht, während das mittlere korpuskuläre Volumen (MCV) der Erythrozyten und/oder das mittlere korpuskuläre Hämoglobin (MCH) vermindert sind. Die Anzahl der roten Blutkörperchen ist in der Regel normal oder erhöht, kann aber verringert sein, wenn der Patient andere Ursachen für die Anämie hat.
Beta-Thalassämie-Trait (auch Beta-Thalassämie Minor oder Beta-Thalassämie-Trägerstatus genannt) ist eine gutartige, heterozygote Erkrankung, die sich durch klinische und labortechnische Merkmale von den schwereren Beta-Thalassämie-Syndromen (Intermedia und Major) unterscheiden lässt. Beta-Thalassämie intermedia und major sind mit einem zunehmenden Schweregrad der Anämie, Transfusionsabhängigkeit und Splenomegalie verbunden, während diese Merkmale bei Beta-Thalassämie trait nicht vorhanden sind. Bei schwerer Beta-Thalassämie ist der Gehalt an fetalem Hämoglobin (Hb F) aufgrund des Fehlens von Hb A deutlich erhöht, und die Menge an Hb A2 ist möglicherweise nicht wie bei Beta-Thalassämie-Trait erhöht. Die Labordiagnose der Beta-Thalassämie sollte also relativ einfach sein, richtig?
Nicht immer. Die folgenden zwei Fälle veranschaulichen häufige Situationen, die die Diagnose erschweren können.
Fall 1: Konkurrierende Bedingungen
Bei der Patientin handelte es sich um eine 28-jährige Afroamerikanerin, die positiv für eine HIV-1-Infektion war und eine hochaktive antiretrovirale Therapie (HAART) mit einem Dolutegravir/Abacavir/Lamivudin-Schema erhielt. Die Laborbefunde sind in Tabelle 1 aufgeführt.
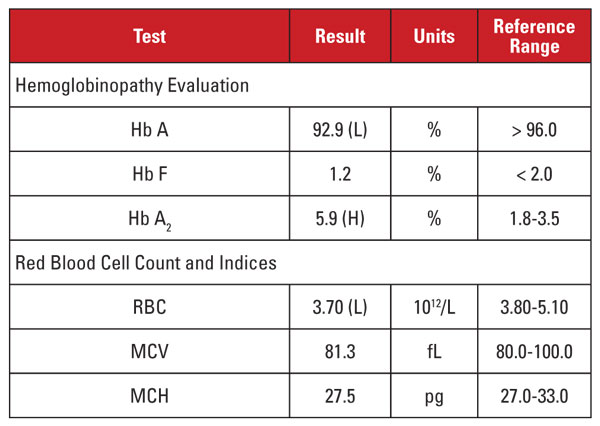
Dieser Fall stellte eine interessante Situation dar. Obwohl der Hb A2-Wert signifikant erhöht war, wies das entsprechende Hämogramm weder eine Mikrozytose noch eine Hypochromie auf. Wenn die Laborwerte nicht mit der Diagnose einer Beta-Thalassämie übereinstimmen, ist es sinnvoll, andere Erkrankungen in Betracht zu ziehen, die den Hb A2-Spiegel und die Erythrozytenindizes beeinflussen. In diesem Fall wurde ein wichtiger Hinweis in der klinischen Vorgeschichte des Patienten gefunden.
Der Hb A2-Spiegel steigt tendenziell bei Erkrankungen, die die Kernreifung der Erythrozytenvorläufer verzögern. Diese Zustände sind auch mit einem erhöhten MCV verbunden.1,2 Die häufigste Ursache für dieses Phänomen ist eine megaloblastische Anämie aufgrund eines Folat- und/oder Vitamin-B12-Mangels. Mehrere Medikamente, die die Nukleinsäuresynthese hemmen, haben jedoch eine ähnliche Wirkung, darunter die Klasse der Anti-HIV-Medikamente, die so genannten Nukleosid-Reverse-Transkriptase-Inhibitoren (NRTI). Lamivudin im HAART-Schema dieses Patienten ist ein NRTI-Medikament.
Sollten wir also zu dem Schluss kommen, dass der hohe Hb A2-Wert dieses Patienten auf die Anti-HIV-Medikamente zurückzuführen ist und der Patient keine Beta-Thalassämie aufweist? Nicht so schnell! Im Allgemeinen ist der Anstieg von Hb A2 aufgrund von NRTI (oder megaloblastischer Anämie) geringer als bei Beta-Thalassämie.1,2 Außerdem sind die Anstiege in der Regel proportional zu den Gesamteffekten des Medikaments, die durch einen Anstieg des MCV angenähert werden können.1,2 Bei vielen Patienten, die eine NRTI-Therapie erhalten, ist das MCV deutlich erhöht (oft über 120 fL), während das MCV bei unserem Patienten eher am unteren Ende des Referenzbereichs lag.
Daher waren wir nicht davon überzeugt, dass der sehr hohe Hb A2-Wert des Patienten ausschließlich auf die Lamivudin-Therapie zurückzuführen war. Wir vermuteten, dass bei dem Patienten konkurrierende Bedingungen vorlagen, die den MCV-Wert erhöhten bzw. verringerten (d. h. Beta-Thalassämie und Lamivudin-Therapie), wobei beide zu einem hohen Hb A2-Wert beitrugen. Wir teilten dem behandelnden Arzt mit, dass eine Beta-Thalassämie wahrscheinlich sei und durch eine Beta-Globin-Mutationsanalyse bestätigt werden könne. Die anschließende Gensequenzierung ergab eine heterozygote Beta-Globin-Mutation, die mit einer Beta-Null-Thalassämie assoziiert war.
Fall 2: Ethnischer Faktor
Die Patientin war eine 35-jährige schwangere Vietnamesin, die auf Hämoglobinopathie untersucht wurde. Die Laborergebnisse sind in Tabelle 2 aufgeführt.
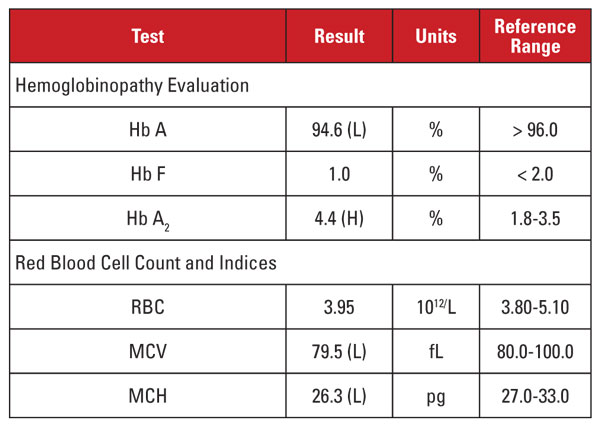
Auch hier war der hohe Hb A2-Wert nicht mit minimaler oder fehlender Mikrozytose und Hypochromie vereinbar. In diesem Fall konnten wir häufige Störfaktoren wie Folat-, Vitamin-B12- oder Eisenmangel, Schilddrüsenfehlfunktionen oder Medikamentenwirkungen ausschließen. Aufgrund der ethnischen Zugehörigkeit des Patienten vermuteten wir eine weitere wichtige Ursache für die Diskordanz: das Beta-Thalassämie-Merkmal, das gemeinsam mit der Alpha-Thalassämie minor vererbt wird. Wenn ein Patient beide Erkrankungen erbt, normalisieren sich MCV und MCH häufig, da Alpha- und Beta-Globinketten in den sich entwickelnden roten Blutkörperchen in relativ ausgeglichenen Mengen vorhanden sind.2
Wir empfahlen genetische Analysen, um auf Alpha- und Beta-Thalassämie-Mutationen zu testen, um das Risiko für die schwangere Mutter und den Fötus vollständig abzuschätzen. Die anschließende Beta-Globin-Sequenzierung ergab eine heterozygote Beta-plus-Thalassämie-Mutation, während die Analyse auf Alpha-Globin-Deletionen eine heterozygote südostasiatische (SEA) Zwei-Gen-Deletion ergab, die mit einer -/αα-Alpha-Thalassämie minor übereinstimmt. Aufgrund dieser Befunde war eine genetische Analyse des Vaters angezeigt, um das Risiko für den Fötus abzuschätzen, ein klinisch schweres Thalassämie-Syndrom zu erben.
Alpha- und Beta-Thalassämien
Thalassämien gehören zu den häufigsten Erbkrankheiten der Menschheit. Sie sind dort weit verbreitet, wo Malaria endemisch war, und kommen heute aufgrund der Migration der menschlichen Bevölkerung in allen Teilen der Welt vor. Thalassämien werden durch Mutationen verursacht, die die Expression des Globin-Gens in den Vorläufern der roten Blutkörperchen verringern. Die Erkrankungen werden nach den mutierten Globin-Genen (z. B. alpha versus beta) und nach der Schwere der Erkrankung klassifiziert, die davon abhängt, ob die Mutationen heterozygot oder homozygot/kompound heterozygot vererbt werden. Die Hauptform des Hämoglobins bei Erwachsenen ist Hb A, ein Tetramer aus zwei Alpha- und zwei Beta-Globinketten. Bei Alpha-Thalassämie ist die Expression von Alpha-Globin mangelhaft und es besteht ein entsprechender Überschuss an Beta-Globin-Ketten.
Bei Beta-Thalassämie ist dieses Muster umgekehrt. Bei einem Mangel an Globinketten und einem Ungleichgewicht ist der Hämoglobingehalt der Erythrozyten verringert, was zu Mikrozytose und Hypochromie führt. Darüber hinaus bilden überschüssige Alpha- oder Beta-Globinketten instabile Tetramere, die eine Hämolyse verursachen. Alpha- und Beta-Thalassämie unterscheiden sich durch die Menge des kleineren Erwachsenenhämoglobins Hb A2, einem Tetramer aus zwei Alpha- und zwei Deltaglobinketten. Hb A2 ist bei Beta-Thalassämie erhöht, da der relative Mangel an Beta-Globin den Einbau von mehr Delta-Ketten in das Hämoglobin ermöglicht.
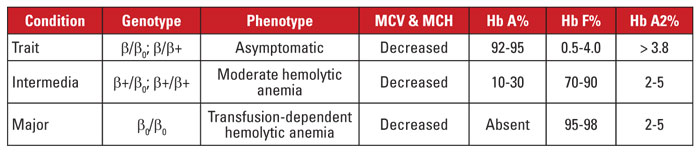
Beta-Thalassämie wird durch Mutationen im Beta-Globin-Genlocus auf Chromosom 11 verursacht.3,4 Bei den meisten Mutationen handelt es sich um kleine Nukleotid-Substitutionen, Insertionen oder Deletionen, obwohl in seltenen Fällen auch große Deletionen festgestellt werden. Je nach Mutation ist die Beta-Globin-Expression teilweise (Beta-Plus-Thalassämie) oder vollständig (Beta-Null-Thalassämie) reduziert. Die Beta-Thalassämie wird durch eine heterozygote Mutation verursacht. Dieser Zustand ist asymptomatisch und zeichnet sich durch einen erhöhten Hb A2, eine Mikrozytose der Erythrozyten und keine signifikante hämolytische Anämie aus. Im Gegensatz dazu wird die Beta-Thalassämie major (Cooley’sche Anämie) durch homozygote Beta-Null-Mutationen verursacht. Die Hämoglobinauswertung zeigt ein Übergewicht von Hb F, fehlendes Hb A und normales oder erhöhtes Hb A2. Die Beta-Thalassämie major ist durch eine schwere, transfusionsabhängige hämolytische Anämie gekennzeichnet, die zu Splenomegalie und Knochendeformitäten führt. Wiederholte Transfusionen führen zu einer Eisenüberladung, die eine der Hauptursachen für Morbidität und Mortalität ist. Die Beta-Thalassämie intermedia ist klinisch heterogen, wobei die meisten Symptome mit einer moderaten hämolytischen Anämie zusammenhängen. Transfusionsabhängigkeit ist ungewöhnlich, da die Beta-Globin-Expression nicht fehlt. Aufgrund der großen Anzahl von Mutationen, die mit der Beta-Thalassämie assoziiert sind, erfordert die genetische Diagnose in der Regel eine Gensequenzierung. Die Beta-Thalassämie-Syndrome sind in Tabelle 3 zusammengefasst.
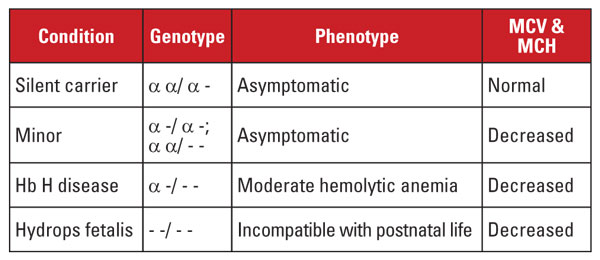
Alpha-Thalassämie wird durch Mutationen im Alpha-Globin-Genlocus auf Chromosom 16 verursacht.5 Alpha-Globin-Gene sind auf Chromosom 16 dupliziert, so dass ein normales Individuum vier Kopien besitzt. Die häufigsten Mutationen sind große Deletionen, die ein oder beide Gene auf dem Locus betreffen. Es wurde eine recht begrenzte Anzahl von Deletionen beschrieben, die in verschiedenen Populationen vorkommen; beispielsweise sind große Mutationen, die beide Alpha-Gene löschen, bei Menschen südostasiatischer Ethnizität sehr häufig. Diese Mutationen sind auch in der mediterranen Bevölkerung weit verbreitet, bei Menschen afrikanischer Abstammung sind sie jedoch selten. Im Gegensatz dazu sind Deletionen einzelner Alpha-Globin-Gene weit verbreitet. Der Verlust eines der vier Alpha-Globin-Gene wird als stiller Trägerzustand bezeichnet.
Dieser Zustand ist asymptomatisch und geht in der Regel mit normalen Erythrozyten-Indizes einher. Der Verlust von zwei Alpha-Globin-Genen wird als Alpha-Thalassämie minor bezeichnet. Dieser Zustand kann durch homozygote Deletionen eines Gens oder durch heterozygote Deletionen von zwei Genen entstehen. Die Erythrozytenzahl und die Erythrozytenindizes sind nicht von der Beta-Thalassämie zu unterscheiden, aber der Hb A2-Wert ist normal. Der Verlust von drei Alpha-Genen wird als Hb H-Krankheit bezeichnet. (Hb H ist ein Tetramer aus Betaglobinketten.) Die Hb H-Krankheit ist typischerweise eine mäßige hämolytische Anämie, die mit Splenomegalie und Knochenveränderungen einhergeht. Transfusionsabhängigkeit ist selten.
Der Verlust aller vier Alpha-Gene wird als Hb Barts-Hydrops fetalis bezeichnet (Hb Barts ist ein Tetramer fetaler Gamma-Globinketten). Dieser Zustand ist mit dem postnatalen Leben unvereinbar. Hb H und Hb Barts können bei Hämoglobinauswertungen identifiziert werden und spielen eine Rolle bei der postnatalen Diagnose der Alpha-Thalassämie.5
Die genetische Diagnose wird in der Regel durch Lücken-Polymerase-Kettenreaktion (PCR) Assays durchgeführt, die auf die gemeinsamen Deletionen abzielen. Die Sequenzierung des Alpha-Globin-Gens ist auch zum Nachweis seltener Mutationen möglich. Die Alpha-Thalassämie-Syndrome sind in Tabelle 4 zusammengefasst.
Wenn Gentests erforderlich sind
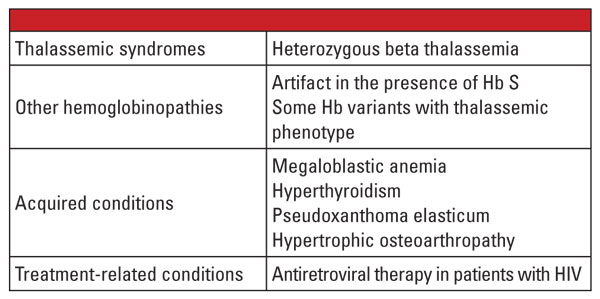
Zusammenfassend lässt sich sagen, dass die heterozygoten Formen der Alpha- und Beta-Thalassämie sehr häufig sind und im klinischen Labor häufig festgestellt werden. Eine Hämoglobinbestimmung und andere klinische und labortechnische Routinedaten reichen in der Regel für die Diagnose aus. Genetische Tests sind in den meisten Fällen nicht erforderlich, können aber im Zusammenhang mit der Familienplanung von entscheidender Bedeutung sein. Wenn bei beiden Elternteilen Thalassämie-Mutationen festgestellt werden, kann für den Fötus ein erhebliches Risiko bestehen, eine schwere Thalassämie zu erben. Im Falle eines Hydrops fetalis ist ein fetales Absterben wahrscheinlich, und für die Mutter besteht ein erhebliches Risiko für Schwangerschaftskomplikationen.5 Daher ist es wichtig, sich der Variablen bewusst zu sein, die die Labordiagnose von Thalassämien erschweren, und zu erkennen, wann ein bestätigender Gentest angezeigt ist. Wie im ersten Fallbericht gezeigt, kann die Diagnose einer Beta-Thalassämie beispielsweise schwierig sein, wenn gleichzeitig Erkrankungen vorliegen, die den Hb A2-Wert beeinflussen. Faktoren, von denen bekannt ist, dass sie Hb A2 erhöhen, sind in Tabelle 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: Biologie, klinische Relevanz und ein mögliches Ziel für die Verbesserung der Sichelzellkrankheit. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Gene test review. Alpha-Thalassemia. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH recommendations for the measurement of haemoglobin A2. Int J Lab Hematol. 2012;34(1):1-13.