I rutinemæssig laboratoriepraksis stilles diagnosen beta thalassemia-trait normalt på baggrund af karakteristiske fund i hæmoglobinevalueringen og antallet af røde blodlegemer og indekser. Navnlig er procentdelen af hæmoglobin (Hb) A2 forhøjet, mens det gennemsnitlige korpuskulære volumen (MCV) af de røde blodlegemer og/eller det gennemsnitlige korpuskulære hæmoglobin (MCH) er nedsat. Antallet af røde blodlegemer er normalt normalt normalt eller forhøjet, men kan være nedsat, hvis patienten har andre årsager til anæmi.
Beta thalassemia trait (også kaldet beta thalassemia minor eller beta thalassemia carrier state) er en godartet, heterozygot tilstand, der kan adskilles fra de mere alvorlige beta thalassemia syndromer (intermedia og major) ved kliniske og laboratoriemæssige karakteristika. Beta thalassemia intermedia og major er forbundet med stigende sværhedsgrad af anæmi, transfusionsafhængighed og splenomegali, mens disse træk ikke forekommer i beta thalassemia trait. Ved svær betathalassæmi er niveauet af føtal hæmoglobin (Hb F) markant forøget på grund af fraværet af Hb A, og mængden af Hb A2 er muligvis ikke forøget som ved betathalassæmitrait. Så laboratoriediagnosen beta thalassemia trait burde være relativt ligetil, ikke sandt?
Ikke altid. De følgende to tilfælde illustrerer almindelige situationer, der kan komplicere diagnosen.
Fald 1: Konkurrerende forhold
Patienten var en 28-årig afroamerikansk kvinde, der var positiv for HIV-1-infektion og var i højaktiv antiretroviral terapi (HAART) med en dolutegravir/abacavir/lamivudin-kur. Laboratoriefund er vist i tabel 1.
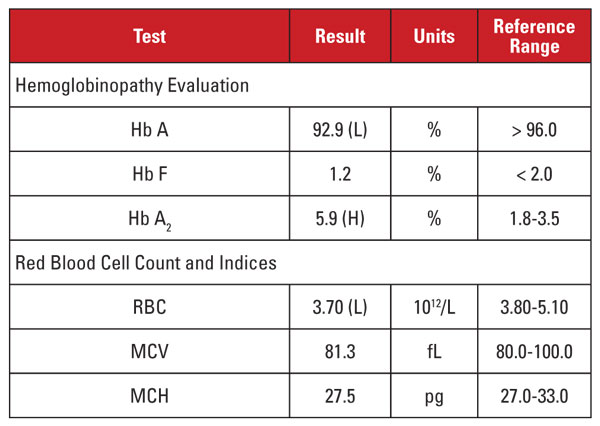
Dette tilfælde præsenterede en interessant situation. Selv om Hb A2-niveauet var betydeligt forhøjet, viste det tilsvarende hæmogram ikke mikrocytose eller hypokromi. Når laboratoriekarakteristika er diskordante for en diagnose af beta thalassemia-trait, er det nyttigt at overveje andre tilstande, der påvirker Hb A2-niveauet og indeks for røde blodlegemer. I dette tilfælde blev der fundet en vigtig ledetråd i patientens kliniske historie.
Hb A2-niveauet har tendens til at stige under forhold, der forsinker kernemodningen af forstadier til røde blodlegemer. Disse tilstande er også forbundet med øget MCV.1,2 Den mest almindelige årsag til dette fænomen er megaloblastisk anæmi som følge af folat- og/eller B12-vitaminmangel. Flere lægemidler, der hæmmer nukleinsyresyntesen, har imidlertid en lignende virkning, herunder den klasse af anti-HIV-medikamenter, der kaldes nukleoside omvendte transkriptasehæmmere (NRTI’er). Lamivudin i denne patients HAART-regime er en NRTI-medicin.
Så skal vi konkludere, at denne patients høje Hb A2-niveau skyldtes anti-HIV-medicinen, og at patienten ikke havde beta thalassemia-træk? Ikke så hurtigt! Generelt er stigninger i Hb A2 på grund af NRTI (eller megaloblastisk anæmi) mindre end det, der ses ved beta thalassemia trait.1,2 Desuden har stigningerne tendens til at være proportionale med de generelle virkninger af lægemidlet, hvilket kan tilnærmes ved stigninger i MCV.1,2 Hos mange patienter i NRTI-behandling er MCV markant høj (ofte større end 120 fL), mens MCV hos vores patient var mod den lave ende af referenceområdet.
Derfor var vi ikke overbevist om, at patientens meget høje Hb A2-niveau udelukkende skyldtes lamivudinbehandling. Vi havde mistanke om, at patienten havde konkurrerende forhold, der øgede og sænkede MCV (dvs. henholdsvis beta thalassemia-træk og lamivudinbehandling), og at begge bidrog til et højt Hb A2-niveau. Vi meddelte den ordinerende læge, at beta thalassemia-træk var sandsynligt, og at det kunne bekræftes ved hjælp af beta globinmutationsanalyse. Efterfølgende gensekventering afslørede en heterozygot betaglobinmutation, der var forbundet med beta-zero thalassæmi.
Fald 2: etnicitetsfaktor
Patienten var en 35-årig gravid vietnamesisk kvinde, der blev screenet for hæmoglobinopati. Laboratoriefundene er vist i tabel 2.
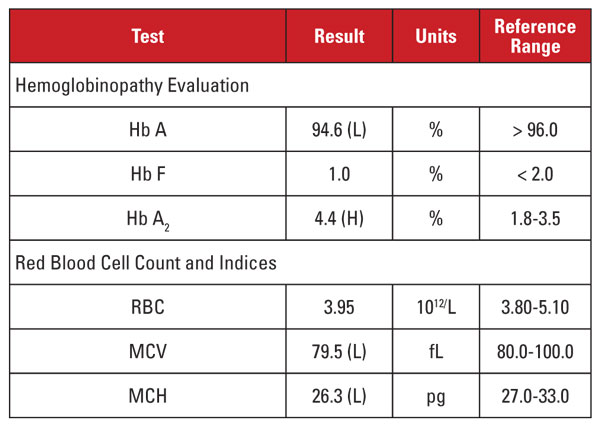
Det høje Hb A2-niveau var igen i uoverensstemmelse med minimal eller fraværende mikrocytose og hypokromi. I dette tilfælde var vi i stand til at udelukke almindelige forvirrende faktorer såsom folat-, B12-vitamin- eller jernmangel, skjoldbruskkirteldysfunktion eller medicinvirkninger. På baggrund af patientens etnicitet mistænkte vi en anden vigtig årsag til diskordans: beta thalassemia trait co-inherited with alpha thalassemia minor. Når en patient arver begge disse tilstande, normaliseres MCV og MCH ofte, fordi alfa- og betaglobinkæderne er til stede i relativt afbalancerede mængder i røde blodlegemer under udvikling.2
Vi anbefalede genetiske analyser for at teste for alfa- og betathalassæmi-mutationer for fuldt ud at kunne vurdere risikoen for den gravide mor og fosteret. Efterfølgende betaglobin-sekventering afslørede en heterozygot beta-plus thalassemia-mutation, mens analyse for alfaglobindeletioner afslørede en heterozygot sydøstasiatisk (SEA) to-gen-deletion, hvilket stemmer overens med -/ααα alpha thalassemia minor. På baggrund af disse fund var genetisk analyse af faderen indiceret for at vurdere risikoen for fosteret for at arve et klinisk alvorligt thalassæmisyndrom.
Alpha- og beta- thalassemier
Thalassemier er blandt de mest almindelige arvelige sygdomme i menneskeheden. De er meget udbredte, hvor malaria har været endemisk, og de er nu almindelige i alle dele af verden på grund af migration af menneskelige befolkninger. Thalassemias skyldes mutationer, der reducerer globin-genekspressionen i forstadier til røde blodlegemer. Sygdommene klassificeres efter de globingener, der er muteret (f.eks. alfa versus beta), og efter sygdommens sværhedsgrad, som afhænger af, om mutationerne nedarves heterozygot eller homozygot/kombineret heterozygot. Den vigtigste voksenform af hæmoglobin er Hb A, en tetramer af to alfa- og to betaglobinkæder. Ved alfa-thalassæmi er alfa-globin-ekspressionen mangelfuld, og der er et tilsvarende overskud af betaglobinkæder.
Dette mønster er omvendt ved beta-thalassæmi. I forbindelse med mangel på og ubalance i globinkæderne er indholdet af hæmoglobin i de røde blodlegemer nedsat, hvilket resulterer i mikrocytose og hypokromi. Desuden danner overskydende alfa- eller betaglobinkæder ustabile tetramere, der forårsager hæmolyse. Man skelner mellem alfa- og beta-talassæmi på grundlag af mængden af det mindre voksne hæmoglobin Hb A2, som er en tetramer af to alfa- og to delta-globinkæder. Hb A2 er øget ved beta-thalassæmi, fordi den relative mangel på betaglobin gør det muligt at inkorporere flere deltakæder i hæmoglobin.
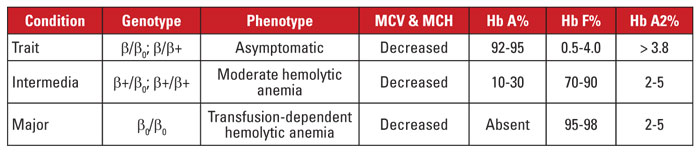
Beta-thalassæmi skyldes mutationer i betaglobin-genlokus på kromosom 11.3,4 De fleste mutationer er små nukleotid-substitutioner, indsættelser eller deletioner, selv om der i sjældne tilfælde identificeres store deletioner. Afhængigt af mutationen er betaglobinekspressionen reduceret delvist (beta-plus thalassæmi) eller helt (beta-zero thalassæmi). Beta thalassemia-træk er forårsaget af en heterozygot mutation. Denne tilstand er asymptomatisk og er karakteriseret ved øget Hb A2, mikrocytose i de røde blodlegemer og ingen betydelig hæmolytisk anæmi. I modsætning hertil skyldes beta thalassemia major (Cooley’s anæmi) homozygote beta-zero-mutationer. Hæmoglobinevalueringen afslører en overvægt af Hb F, manglende Hb A og normal eller forhøjet Hb A2. Beta thalassemia major er karakteriseret ved alvorlig, transfusionsafhængig hæmolytisk anæmi med deraf følgende splenomegali og knogledeformiteter. Tilbagevendende transfusioner fører til jernoverbelastning, som er en hovedårsag til morbiditet og mortalitet. Beta thalassemia intermedia er klinisk heterogen, og de fleste symptomer er relateret til moderat hæmolytisk anæmi. Transfusionsafhængighed er ualmindelig, fordi betaglobinudtryk ikke er fraværende. På grund af det store antal mutationer, der er forbundet med beta thalassæmi, kræver genetisk diagnose typisk sekventering af generne. Beta thalassemia syndromerne er opsummeret i tabel 3.
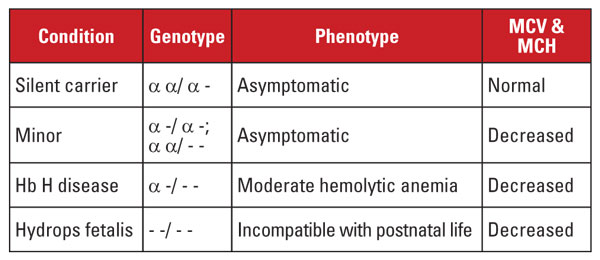
Alpha thalassæmi skyldes mutationer i alfa globin-genlokus på kromosom 16.5 Alfa globin generne er duplikeret på kromosom 16, så et normalt individ har fire kopier. De mest almindelige mutationer er store deletioner, der påvirker et eller begge gener på locus. Der er beskrevet et ret begrænset antal deletioner med prævalens i forskellige befolkningsgrupper; f.eks. er store mutationer, der sletter begge alfa-gener, meget almindelige hos personer af sydøstasiatisk etnisk oprindelse. Disse mutationer er også udbredt i middelhavspopulationer, men er sjældne hos mennesker af afrikansk etnisk oprindelse. I modsætning hertil er deletioner af enkelte alfa-globingener meget udbredt. Tabet af et af de fire alfa-globingener betegnes som en silent carrier state.
Denne tilstand er asymptomatisk og er normalt forbundet med normale indekser for røde blodlegemer. Tab af to alfa-globingener betegnes alfa-thalassæmi minor. Denne tilstand kan forekomme på grund af homozygote enkeltgen-deletioner eller heterozygote togen-deletioner. Antallet af røde blodlegemer og indeks er ikke til at skelne fra beta thalassemia trait, men Hb A2-niveauet er normalt. Tabet af tre alfa-gener betegnes Hb H-sygdom. (Hb H er en tetramer af betaglobinkæder.) Hb H-sygdom er typisk en moderat hæmolytisk anæmi, der er forbundet med splenomegali og knogleforandringer. Transfusionsafhængighed er ualmindeligt.
Tabet af alle fire alfa-gener betegnes Hb Barts hydrops fetalis (Hb Barts er en tetramer af føtale gammaglobinkæder). Denne tilstand er uforenelig med det postnatale liv. Hb H og Hb Barts kan identificeres ved hæmoglobinvurderinger og spiller en rolle i den postnatale diagnose af alfa-thalassæmi.5
Genetisk diagnose foretages normalt ved hjælp af gap-polymerasekædereaktions-assays (PCR), der er rettet mod de almindelige deletioner. Der findes også sekventering af alfa-globin-genet for at påvise sjældne mutationer. Alpha thalassemia syndromerne er opsummeret i tabel 4.
Når der er behov for genetisk testning
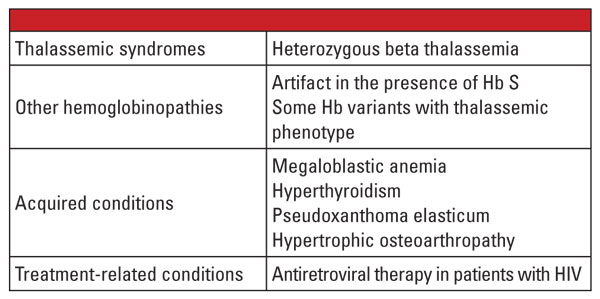
Sammenfattende er de heterozygote former af alfa- og beta- thalassæmi meget almindelige og identificeres hyppigt i det kliniske laboratorium. Hæmoglobinevaluering og andre rutinemæssige kliniske og laboratoriedata er normalt tilstrækkelige til at stille diagnosen. Genetisk testning er ikke nødvendig i de fleste tilfælde, men kan være af afgørende betydning i forbindelse med familieplanlægning. Hvis der identificeres thalassemia-mutationer hos begge forældre, kan der være en betydelig risiko for, at fosteret arver alvorlig thalassæmi. I tilfælde af hydrops fetalis er der sandsynlighed for fosterdød, og der er en betydelig risiko for moderen for graviditetskomplikationer.5 Derfor er det vigtigt at være opmærksom på variabler, der komplicerer laboratoriediagnosen af thalassemier, og erkende, hvornår bekræftende genetisk test er indiceret. For eksempel kan det, som det fremgår af den første case report, være en udfordring at stille diagnosen beta thalassemia trait, hvis der er sameksisterende tilstande, der påvirker niveauet af Hb A2. Faktorer, der er kendt for at øge Hb A2, er opført i tabel 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biologi, klinisk relevans og et muligt mål for forbedring af seglcellesygdom. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Gennemgang af gentest. Alpha-thalassæmi. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH-anbefalinger for måling af hæmoglobin A2. Int J Lab Hematol. 2012;34(1):1-13.