Dans la pratique de laboratoire de routine, le diagnostic du trait de bêta-thalassémie est généralement posé par des résultats caractéristiques dans l’évaluation de l’hémoglobine et la numération et les indices des globules rouges. En particulier, le pourcentage d’hémoglobine (Hb) A2 est élevé, tandis que le volume corpusculaire moyen (MCV) et/ou l’hémoglobine corpusculaire moyenne (MCH) des globules rouges sont diminués. La numération des globules rouges est généralement normale ou augmentée, mais peut être diminuée si le patient présente d’autres causes d’anémie.
Le trait de bêta-thalassémie (également appelé bêta-thalassémie mineure ou état de porteur de bêta-thalassémie) est une condition hétérozygote bénigne qui peut être distinguée des syndromes de bêta-thalassémie plus sévères (intermédia et majeur) par des caractéristiques cliniques et de laboratoire. Les syndromes intermedia et major sont associés à une gravité croissante de l’anémie, à une dépendance aux transfusions et à une splénomégalie, alors que ces caractéristiques sont absentes du trait de bêta-thalassémie. Dans la bêta-thalassémie sévère, le taux d’hémoglobine fœtale (Hb F) est nettement plus élevé en raison de l’absence d’Hb A, et la quantité d’Hb A2 peut ne pas être augmentée comme dans le cas du trait de bêta-thalassémie. Le diagnostic de laboratoire du trait de bêta-thalassémie devrait donc être relativement simple, n’est-ce pas ?
Pas toujours. Les deux cas suivants illustrent des situations courantes qui peuvent compliquer le diagnostic.
Cas 1 : conditions concurrentes
La patiente était une Afro-Américaine de 28 ans qui était positive à l’infection par le VIH-1 et suivait une thérapie antirétrovirale hautement active (HAART) avec un régime dolutegravir/abacavir/lamivudine. Les résultats de laboratoire sont présentés dans le tableau 1.
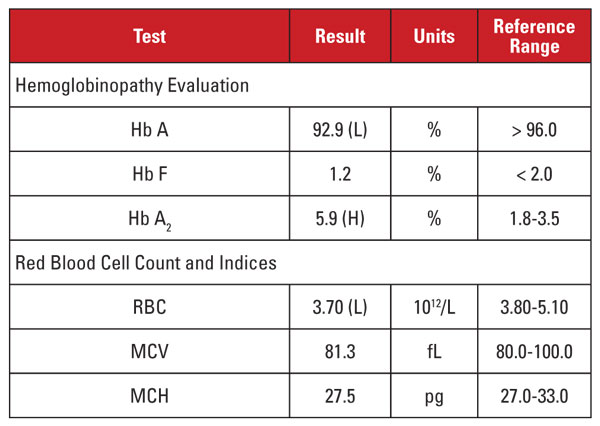
Ce cas présentait une situation intéressante. Bien que le taux d’Hb A2 ait été significativement élevé, l’hémogramme correspondant ne démontrait pas de microcytose ou d’hypochromie. Lorsque les caractéristiques de laboratoire sont discordantes pour un diagnostic de trait de bêta-thalassémie, il est utile de considérer d’autres conditions qui affectent le taux d’Hb A2 et les indices de globules rouges. Dans ce cas, un indice important a été trouvé dans l’histoire clinique du patient.
Les taux d’Hb A2 ont tendance à augmenter dans les conditions qui retardent la maturation nucléaire des précurseurs des globules rouges. Ces conditions sont également associées à une augmentation du VCM.1,2 La cause la plus courante de ce phénomène est l’anémie mégaloblastique due à une carence en folates et/ou en vitamine B12. Cependant, plusieurs médicaments qui inhibent la synthèse des acides nucléiques ont un effet similaire, notamment la classe de médicaments anti-VIH appelés inhibiteurs nucléosidiques de la transcriptase inverse (INTI). La lamivudine dans le régime HAART de ce patient est un médicament NRTI.
Donc, devons-nous conclure que le taux élevé d’Hb A2 de ce patient était dû au médicament anti-VIH, et que le patient n’avait pas le trait de bêta-thalassémie ? Pas si vite ! En général, les augmentations du taux d’Hb A2 dues aux INTI (ou anémie mégaloblastique) sont moins importantes que celles observées dans le cas du trait de bêta-thalassémie.1,2 De plus, les augmentations ont tendance à être proportionnelles aux effets globaux du médicament, qui peuvent être approximés par des augmentations du VCM.1,2 Chez de nombreux patients traités par des INTI, le VCM est nettement élevé (souvent supérieur à 120 fL), alors que chez notre patient, le VCM se situait vers l’extrémité inférieure de la plage de référence.
Par conséquent, nous n’étions pas convaincus que le taux très élevé d’Hb A2 de notre patient était uniquement dû au traitement par la lamivudine. Nous soupçonnions que le patient avait des conditions concurrentes qui augmentaient et diminuaient le VCM (c’est-à-dire le trait de bêta-thalassémie et le traitement à la lamivudine, respectivement), les deux contribuant à un taux élevé d’Hb A2. Nous avons signalé au médecin traitant qu’un trait de bêta-thalassémie était probable et qu’il pouvait être confirmé par une analyse de la mutation de la bêta-globine. Le séquençage génétique ultérieur a révélé une mutation hétérozygote de la bêta-globine associée à la bêta-thalassémie.
Cas 2 : facteur ethnique
La patiente était une femme vietnamienne enceinte de 35 ans qui faisait l’objet d’un dépistage de l’hémoglobinopathie. Les résultats de laboratoire sont présentés dans le tableau 2.
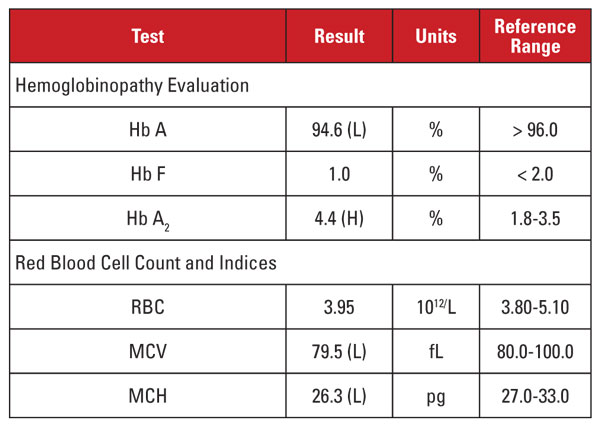
De nouveau, le taux élevé d’Hb A2 était discordant avec une microcytose et une hypochromie minimes ou absentes. Dans ce cas, nous avons pu exclure les facteurs de confusion courants tels que la carence en folates, en vitamine B12 ou en fer, le dysfonctionnement de la thyroïde ou les effets des médicaments. Sur la base de l’origine ethnique du patient, nous avons suspecté une autre cause importante de discordance : le trait de bêta-thalassémie co-inhérité avec l’alpha-thalassémie mineure. Lorsqu’un patient hérite de ces deux conditions, le VCM et l’HME se normalisent souvent parce que les chaînes de globine alpha et bêta sont présentes en quantités relativement équilibrées dans les globules rouges en développement.2
Nous avons recommandé des analyses génétiques pour tester les mutations de l’alpha et de la bêta thalassémie, afin d’évaluer pleinement le risque pour la mère enceinte et le fœtus. Le séquençage ultérieur de la bêta-globine a révélé une mutation hétérozygote de la thalassémie bêta-plus, tandis que l’analyse des délétions de l’alpha-globine a révélé une délétion hétérozygote de deux gènes de l’Asie du Sud-Est (ASE), compatible avec une alpha-thalassémie mineure -/αα. Sur la base de ces résultats, une analyse génétique du père était indiquée pour évaluer le risque pour le fœtus d’hériter d’un syndrome de thalassémie cliniquement sévère.
Alpha et bêta thalassémies
Les thalassémies sont parmi les troubles héréditaires les plus courants dans l’humanité. Elles sont très répandues là où le paludisme a été endémique, et sont maintenant courantes dans toutes les parties du monde en raison de la migration des populations humaines. Les thalassémies sont causées par des mutations qui réduisent l’expression du gène de la globine dans les précurseurs des globules rouges. Les troubles sont classés en fonction des gènes de la globine qui sont mutés (par exemple, alpha ou bêta) et de la gravité de la maladie, qui est liée au fait que les mutations sont héritées de façon hétérozygote ou homozygote/hétérozygote composite. La principale forme d’hémoglobine chez l’adulte est l’Hb A, un tétramère composé de deux chaînes de globine alpha et de deux chaînes de globine bêta. Dans l’alpha thalassémie, l’expression de l’alpha globine est déficiente et il existe un excès correspondant de chaînes de bêta globine.
Ce schéma est inversé dans la bêta thalassémie. Dans le cadre d’une déficience et d’un déséquilibre des chaînes de globine, le contenu en hémoglobine des globules rouges est diminué, ce qui entraîne une microcytose et une hypochromie. En outre, les chaînes de globine alpha ou bêta en excès forment des tétramères instables qui provoquent une hémolyse. Les thalassémies alpha et bêta se distinguent par la quantité d’hémoglobine mineure adulte Hb A2, un tétramère de deux chaînes de globine alpha et deux chaînes de globine delta. L’Hb A2 est augmentée dans la bêta thalassémie parce que l’absence relative de bêta globine permet d’incorporer plus de chaînes delta dans l’hémoglobine.
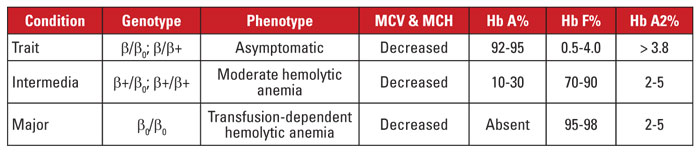
La bêta thalassémie est causée par des mutations dans le locus du gène de la bêta globine sur le chromosome 11.3,4 La plupart des mutations sont de petites substitutions, insertions ou délétions de nucléotides, bien que de grandes délétions soient identifiées dans de rares cas. Selon la mutation, l’expression de la bêta-globine est réduite partiellement (thalassémie bêta-plus) ou complètement (thalassémie bêta-zéro). Le trait de la bêta-thalassémie est causé par une mutation hétérozygote. Cette condition est asymptomatique et se caractérise par une augmentation de l’Hb A2, une microcytose des globules rouges et aucune anémie hémolytique significative. En revanche, la bêta-thalassémie majeure (anémie de Cooley) est due à des mutations homozygotes bêta-zéro. L’évaluation de l’hémoglobine révèle une prédominance de l’Hb F, une absence d’Hb A et une Hb A2 normale ou accrue. La bêta-thalassémie majeure se caractérise par une anémie hémolytique grave, dépendante des transfusions, avec pour conséquence une splénomégalie et des déformations osseuses. Les transfusions récurrentes entraînent une surcharge en fer, qui est une cause principale de morbidité et de mortalité. La bêta-thalassémie intermédiaire est cliniquement hétérogène, la plupart des symptômes étant liés à une anémie hémolytique modérée. La dépendance aux transfusions est peu fréquente car l’expression de la bêta-globine n’est pas absente. En raison du grand nombre de mutations associées à la bêta-thalassémie, le diagnostic génétique nécessite généralement le séquençage du gène. Les syndromes de bêta-thalassémie sont résumés dans le tableau 3.
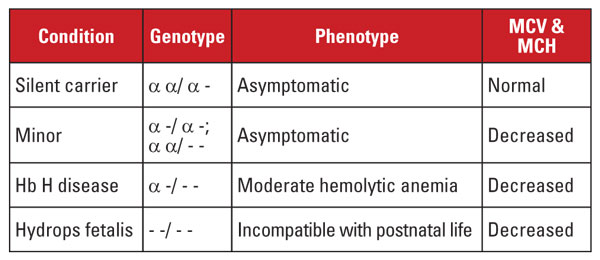
La bêta-thalassémie est causée par des mutations dans le locus du gène de l’alpha globine sur le chromosome 16.5 Les gènes de l’alpha globine sont dupliqués sur le chromosome 16, de sorte qu’un individu normal possède quatre copies. Les mutations les plus courantes sont de grandes délétions qui affectent un ou les deux gènes du locus. Un nombre assez limité de délétions a été décrit, avec une prévalence dans différentes populations ; par exemple, les grandes mutations qui suppriment les deux gènes alpha sont très fréquentes chez les personnes d’origine asiatique du Sud-Est. Ces mutations sont également répandues dans les populations méditerranéennes, mais sont rares chez les personnes d’origine africaine. En revanche, les délétions d’un seul gène alpha-globine sont largement répandues. La perte d’un des quatre gènes de l’alpha-globine est qualifiée d’état de porteur silencieux.
Cette condition est asymptomatique et est généralement associée à des indices normaux de globules rouges. La perte de deux gènes de l’alpha-globine est appelée alpha-thalassémie mineure. Cette affection peut être due à une délétion homozygote d’un gène ou à une délétion hétérozygote de deux gènes. La numération et les indices érythrocytaires sont indiscernables du trait de la bêta-thalassémie, mais le taux d’Hb A2 est normal. La perte de trois gènes alpha est appelée la maladie de Hb H. (L’Hb H est un tétramère de chaînes de globine bêta.) La maladie de l’Hb H est typiquement une anémie hémolytique modérée associée à une splénomégalie et à des modifications osseuses. La dépendance aux transfusions est rare.
La perte des quatre gènes alpha est appelée hydrops fetalis Hb Barts (Hb Barts est un tétramère de chaînes de gammaglobine fœtale). Cette condition est incompatible avec la vie postnatale. L’Hb H et l’Hb Barts peuvent être identifiées lors des évaluations de l’hémoglobine et jouent un rôle dans le diagnostic postnatal de l’alpha thalassémie.5
Le diagnostic génétique est généralement réalisé par des tests de réaction en chaîne par polymérase (PCR) de gap qui ciblent les délétions communes. Le séquençage du gène de l’alpha globine est également disponible pour détecter les mutations rares. Les syndromes de l’alpha thalassémie sont résumés dans le tableau 4.
Quand un test génétique est nécessaire
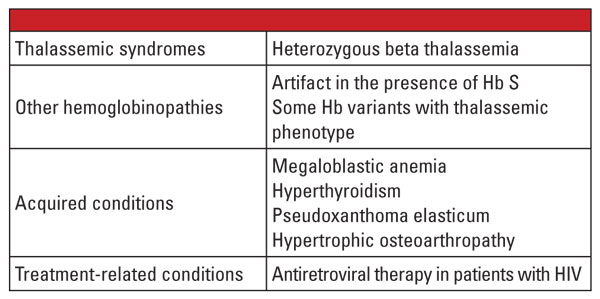
En conclusion, les formes hétérozygotes d’alpha et de bêta thalassémie sont très fréquentes et sont identifiées fréquemment au laboratoire clinique. L’évaluation de l’hémoglobine et d’autres données cliniques et de laboratoire de routine sont généralement suffisantes pour le diagnostic. Le test génétique n’est pas nécessaire dans la plupart des cas, mais peut être d’une importance capitale dans le contexte du planning familial. Si des mutations de la thalassémie sont identifiées chez les deux parents, il peut y avoir un risque important pour le fœtus d’hériter d’une thalassémie grave. Dans le cas d’une anasarque, la mort du fœtus est probable et la mère court un risque important de complications de la grossesse.5 Il est donc important de connaître les variables qui compliquent le diagnostic de laboratoire des thalassémies et de savoir quand un test génétique de confirmation est indiqué. Par exemple, comme le montre le premier rapport de cas, le diagnostic du trait de bêta-thalassémie peut être difficile s’il existe des conditions coexistantes qui affectent le niveau d’Hb A2. Les facteurs connus pour augmenter l’Hb A2 sont énumérés dans le tableau 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin : Genetics, Pathophysiology Clinical Management. Cambridge : Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2 : biologie, pertinence clinique et une cible possible pour améliorer la drépanocytose. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Bêta-thalassémie. Genet Med. 2010;12(2):61-76.
- Origa R. β-Thalassémie. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Revue des tests génétiques. Alpha-thalassémie. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. Recommandations de l’ICSH pour la mesure de l’hémoglobine A2. Int J Lab Hematol. 2012;34(1):1-13.