Abstrakt
Wprowadzenie: Anoftalmia wrodzona to kliniczny brak oka po urodzeniu. Wynika on z braku rozwoju lub regresji pierwotnego pęcherzyka wzrokowego podczas życia embrionalnego. Występuje rzadko i może być izolowana lub związana z innymi wadami ocznymi lub ogólnymi wadami wrodzonymi.
Obserwacje: Opisujemy przypadki trojga dzieci z tej samej rodziny w wieku od 4 miesięcy do 8 lat z obustronną wrodzoną anoftalmią. Ich historia charakteryzuje się brakiem konsultacji i prenatalnych badań laboratoryjnych. Badanie okulistyczne zobiektywizowało, otwierając powieki dwojgiem oczu, wypełnienie ubytku przez spojówkę w 3 przypadkach sugerujące obustronną anoftalmię, potwierdziło USG oczu.
Dyskusja: Anophthalmia wrodzona jest rzadko występującą wadą rozwojową. Może występować pojedynczo lub w zespole wad rozwojowych. Obraz kliniczny jest najczęściej jednostronny. Rozpoznanie jest głównie kliniczne i potwierdzone badaniem ultrasonograficznym gałki ocznej. Przyczyny są różnorodne, reprezentowane przez aberracje chromosomalne, mutacje genetyczne, zatrucia i infekcje nabyte w czasie ciąży.
Wnioski: Stwierdzenie wrodzonej anoftalmii narzuca konieczność przeprowadzenia kompleksowego przeglądu w celu poszukiwania etiologii. Ważnym aspektem pozostaje również wsparcie psychologiczne rodziców.
Słowa kluczowe
anoftalmia, wrodzona, USG gałki ocznej, aberracje chromosomalne
Wprowadzenie
Antalmia jest klinicznym brakiem oka. Jest to niezwykle rzadka nieprawidłowość oka. Istnieje wiele czynników etiologicznych, takich jak aberracje chromosomalne, mutacje genetyczne, zatrucia i infekcje. Anoftalmia wrodzona może być izolowana lub towarzyszyć ciężkim zespołom polimorficznym. Jest ona odpowiedzialna z jednej strony za mikro-orbit i homolateralną hemi-atrofię czaszkowo-twarzową, a z drugiej strony za słaby rozwój wyrostków, co stwarza problemy podczas adaptacji protetycznej. Diagnostyka została udoskonalona dzięki prenatalnym badaniom obrazowym, a postępowanie terapeutyczne polega głównie na stosowaniu protez oczodołu. Przedstawiamy 3 przypadki obustronnej wrodzonej anoftalmii w tej samej rodzinie.
Przypadek 1
Jest to dziecko płci męskiej w wieku 8 lat, urodzone o czasie, przyjęte do konsultacji z powodu obustronnego obniżenia ostrości wzroku. Biologiczna i oczodołowo-mózgowa CT-scan ocena prenatalna nie była możliwa. Kliniczne badanie okulistyczne wykazało brak percepcji świetlnej w obu oczach; obustronną anoftalmię; wyrostki robaczkowe są obecne i prawidłowe, a przy otwarciu powiek widoczna jest struktura spojówkowa wypełniająca całą jamę oczodołową bez gałki ocznej. Zlecona tomografia komputerowa oczodołowo-mózgowa potwierdziła brak globusa ocznego z obecnością resztek tkanki tłuszczowej, mięśni okoruchowych oraz zanik kalibru nerwu wzrokowego w jamie oczodołowej (ryc. 1). Bilans ogólny był prawidłowy. Rezonans magnetyczny nie mógł być wykonany z uwagi na trudności finansowe rodziców, jak również z uwagi na bilans genetyczny (kariotypowanie). Zaproponowano leczenie chirurgiczne z umieszczeniem protezy, ale nie zostało ono zrealizowane z powodu braku odpowiedniej platformy technicznej. (Ryc. 2)
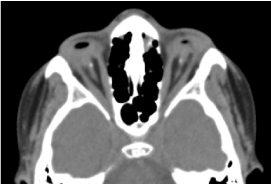
Ryc. 1. Tomografia komputerowa mózgu oczodołu: brak globusa ocznego z resztkami tłuszczu, mięśni i zanikiem kalibru nerwu otokowo-szyjnego w jamie oczodołu.

Rysunek. 2. 8-letnie dziecko z obustronnym wytrzeszczem przy otwieraniu powiek
Przypadek 2
To było 8-letnie dziecko płci męskiej, u którego nie przeprowadzono oceny prenatalnej (biologicznej i ultrasonograficznej). Został on przyjęty do konsultacji okulistycznej z powodu obustronnego obniżenia ostrości wzroku. W klinicznym badaniu okulistycznym stwierdzono brak percepcji świetlnej w obu oczach, obustronną anoftalmię, wyrostki bez cech szczególnych, a przy otwarciu powiek strukturę spojówki wypełniającą ubytek).Wykonana na zlecenie tomografia komputerowa oczodołowo-mózgowa potwierdziła brak globusa ocznego z obecnością resztek tłuszczu, mięśni okulomotorycznych i zanik kalibru nerwu wzrokowego w jamie oczodołowej (ryc. 3). Badanie ogólne dziecka było prawidłowe. Rezonans magnetyczny oraz kariotyp nie mogły być wykonane z tych samych powodów, o których była mowa wcześniej, podobnie jak postępowanie lekarskie. (Ryc. 4)
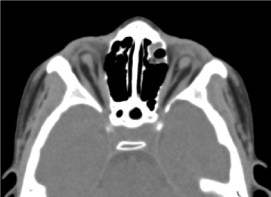
Ryc. 3Brak płata ocznego z resztkami tłuszczu, mięśni i zanikiem kalibru nerwu otokowo-nosowego w jamie oczodołowej.
Przypadek 3
Dotyczy 4-miesięcznego niemowlęcia płci żeńskiej, urodzonego w terminie, o dobrym rozwoju psychomotorycznym i prawidłowej staturo-wadze, które zostało przyjęte na konsultację okulistyczną z powodu obustronnego obniżenia ostrości wzroku i trwałego zamknięcia powiek od urodzenia. W historii niemowlęcia odnotowano dwie konsultacje prenatalne, ale bez prenatalnych badań biologicznych i ultrasonograficznych. W klinicznym badaniu oftalmologicznym stwierdzono brak śledzenia światła; obustronną anoftalmię; normalne aspekty przydatków i do otwierania powiek, ślepy glob oczny dający mikroftalmiczny wygląd, szczegóły tęczówki nie są możliwe do przeanalizowania i brak komory przedniej. Konsultacja pediatryczna w poszukiwaniu innych malformacji powróciła bez żadnych szczegółów na poziomie klinicznym.
Wymagana tomografia oczodołowo-mózgowa potwierdziła brak globusa ocznego z obecnością resztek tłuszczu, mięśni okulomotorycznych i atrofii kalibru nerwu wzrokowego w jamie oczodołowej (rysunek 5). Rezonans magnetyczny i kariotypowanie nie mogły być wykonane; podobnie jak postępowanie medyczne (Rycina 6).

Ryc. 4. 6-letnie dziecko z obustronną anoftalmią przy otwieraniu powiek.
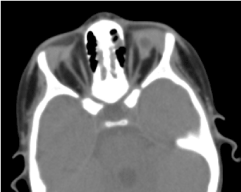
Ryc. 5. Brak oczodołu z resztkami pęcherzyka, mięśni i zanikiem nerwu otokowo-nosowego w jamie oczodołowej.

Ryc.6. 4 miesięczne dziecko; przy otwarciu powiek można było zauważyć, szczególnie w prawym oku, szkic rogówki, ale tęczówka nie jest możliwa do wyodrębnienia.
Dyskusja
Wrodzona anoftalmia lub kliniczny brak oka przy urodzeniu jest rzadką wadą rozwojową. Częstość jej występowania jest zmienna. Szacuje się ją na mniej niż jeden przypadek na 10000 urodzeń dla Mouriaux i Roth . Byłoby to około 21,34 na 100 000 urodzeń zgodnie z hiszpańskim badaniem i 10 na 100 000 urodzeń zgodnie z brytyjskim badaniem . Dla Aranjo , jego częstość występowania wynosi 0,6 na 10000 dzieci urodzonych żywych. Ta różnica w częstości występowania znajduje swoje wyjaśnienie w zmienności występowania tej wady rozwojowej. Na częstość występowania anoftalmii nie ma wpływu rasa ani płeć. Anophthalmia może być izolowana lub włączona do zespołu polimalformacyjnego w jednej trzeciej przypadków. Ta obserwacja jest zgodna z obserwacjami kilku autorów. Tak więc, w przypadku Verny, zarówno anoftalmia jak i mikroftalmia mogą występować w izolacji lub być zintegrowane z zespołem Patau (trisomia13) lub innymi zespołami w około jednej trzeciej przypadków. Kouassi opisał również przypadek obustronnej wrodzonej anoftalmii w przebiegu zespołu Patau. Tak samo Diomandé w Bouaké, który opisał przypadek prawdziwej wrodzonej anoftalmii związanej z ciężką mikroftalmią u noworodka bez żadnych innych obserwowanych nieprawidłowości klinicznych.
W dodatku, Stoll zauważył, że 90% anoftalmii miało towarzyszące wady rozwojowe. Dlatego w obecności jakiejkolwiek wrodzonej anoftalmii lub skrajnej mikroftalmii, ważne jest, aby przeprowadzić dokładną ocenę pediatryczną w poszukiwaniu towarzyszących nieprawidłowości, czasami poważnych, takich jak anencefalia lub agenezja całego układu wzrokowego. Kliniczna anoftalmia rzadko jest obustronna. Nowoczesne badania parakliniczne, takie jak tomografia komputerowa i rezonans magnetyczny, pozwalają na zbadanie towarzyszących im wad rozwojowych. W naszych obserwacjach, te bilanse nie mogły być zrealizowane, biorąc pod uwagę społeczno-ekonomiczne trudności rodziców.
Według Speeg – Schatz , istnieją trzy typy wrodzonej anoftalmii: pierwotna anoftalmia, wtórny anoftalmia i następcza wtórna anoftalmia w wyniku braku inwazji dołu wzrokowego, całkowitego zaprzestania rozwoju przedniego cewy nerwowej lub degeneracji pęcherzyka wzrokowego po jego inwazji. Dla Strickera wrodzona anoftalmitia może być analizowana i klasyfikowana według dwóch sposobów.
Z anatomicznego punktu widzenia należałoby odróżnić prawdziwą anoftalmię od embriopatii charakteryzujących się brakiem jakiegokolwiek zarysu oka. Tylko badanie histologiczne mogłoby to potwierdzić.
Z klinicznego punktu widzenia, kliniczna anoftalmia lub skrajna mikroftalmia musi być oddzielona od innych bardziej umiarkowanych mikroftalmii z czasami lepszym funkcjonalnym rokowaniem. W późniejszych przypadkach występuje bardziej lub rudymentarna gałka oczna (jak w naszych 3. przypadkach klinicznych), której czasami towarzyszy torbiel kolobomatyczna mogąca pełnić rolę fizjologicznej protezy ekspansywnej. Klinicznie nie ma struktury ocznej, ale USG oka, tomografia komputerowa i rezonans magnetyczny wykazałyby obecność szkicu gałki ocznej i nerwu wzrokowego.
Jednakże Morax zauważył, że prawdziwa anoftalmia nie istnieje. Według niego w najbardziej zaznaczonych przypadkach klinicznych, neuroobrazowanie wykazałoby obecność nerwu wzrokowego i maleńkiego szkicu gałki ocznej. W naszych dwóch pierwszych przypadkach klinicznych, USG oka wykazało brak szkicu ocznego. Jednak w przypadku braku wyników tomografii komputerowej i rezonansu magnetycznego nie możemy formalnie stwierdzić, że nie ma nerwu wzrokowego i nerwu wzrokowego.Etiologia anophthalmii jest złożona i różnorodna. Może być dziedziczna poprzez aberracje chromosomalne, w szczególności trisomię 13 lub trisomię 18. W tym przypadku jest to anoftalmia z malformacjami mózgowymi lub związana z dysplazją linii pośrodkowej. Inne etiologie genetyczne są reprezentowane przez mutacje chromosomalne, które mogą wystąpić podczas delecji chromosomalnych lub niezależnie od aberracji kariotypu. Główną z nich są anomalie SOX2. Fantes rozpoznał mikrodelecję związaną z translokacją t (3; 11) (q26-3 p11-2) i mutacją genu SOX2 u pacjenta z obustronną anoftalmią. Uważa się, że haploinsufficiency genu SOX2 jest odpowiedzialny za anoftalmię, mikroftalmię, dysplazję septo-optyczną i defekt w rozwoju przedniej części mózgu. Ye zauważył, że defekt SOX2 spowodowałby liczne zaburzenia układu organicznego, w tym anoftalmię i mikroftalmię. Inne geny, takie jak PAX6, OTX2, CHV10, RAX2, SIX6, NF1, TWIST, SHH, BMP4 i BMP7, byłyby zaangażowane w występowanie anoftalmii. Według Schiltera, mniej niż 40% znanych mutacji genetycznych mogłoby tłumaczyć anoftalmię. Dla niego mutacje genów NF1, PAX6, SOX2 byłyby odpowiedzialne za anoftalmię lub mikroftalmię. Opisała Pani mutację genu OTX2 związaną z anoftalmią i mikroftalmią wrodzoną w chińskiej rodzinie. Weronika zaobserwowała u jednego pacjenta mutację genu RAX2 odpowiedzialną za jednostronną anoftalmię. Mutacje w genie TWIST podczas delecji chromosomu 7 mogą prowadzić do dysgenezji twarzy z anoftalmią. Identyfikacja mutacji allelu genu SIX6 u pacjenta z obustronną anoftalmią została odnotowana przez Gallardo .
Bez kariotypowania nie jesteśmy w stanie przypisać tych dziedzicznych anoftalmii w naszym badaniu do konkretnego typu mutacji. Co więcej, zespół Patau, który jest jedną z głównych etiologii, nie może być zachowany klinicznie u naszych pacjentów.
Z tych wszystkich czynników etiologicznych, teoria mutacji genetycznej wydaje się pasować do naszych przypadków klinicznych, rzeczywiście, te 3 przypadki pochodzą z tej samej rodziny, ale także ze sprzężenia pokrewieństwa, ponieważ oni sami pochodzą z tej samej rodziny, ponieważ są kuzynami. Należy jednak zauważyć, że niedobór witaminy A, ekspozycja na promieniowanie rentgenowskie, nadużywanie rozpuszczalnika, przyjmowanie talidomin i infekcje nabyte w czasie ciąży byłyby również czynnikami środowiskowymi. Diagnoza anophthalmia może być wykonane pre- lub post-natal na podstawie kombinacji objawów klinicznych, obrazowania i analizy genetycznej. Dufit , wykorzystując nowe techniki obrazowania (MRI płodu) opisał przypadek anoftalmii płodowej związanej z licznymi wadami rozwojowymi, co doprowadziło do przerwania ciąży.
W przypadku dzieci leczenie protetyczne polegające na zwiększaniu rozmiarów konformerów, a nawet techniki rozszerzania oczodołów za pomocą nadmuchiwanych protez, jest zalecane przed ukończeniem 2 roku życia. Jednak u dorosłych tego typu leczenie staje się zbędne i odradzane. Schemat leczenia musi obejmować powiększenie kostne oczodołu przez różne osteotomie połączone jednocześnie lub wtórnie z odtworzeniem worka spojówkowego przez przeszczep śluzówkowy lub skórno-naskórkowy. Następnie, odtworzenie powiek następuje poprzez rekonstrukcję, repozycję i korektę nieprawidłowej pozycji na protezie. W naszych krajach rozwijających się, ten rodzaj ciężkiego i czasami rozczarowującego leczenia jest trudny do osiągnięcia przez czasami niewyszkolone zespoły. Co więcej, poziom ubóstwa rodziców ogranicza ich dostępność do leczenia, z konsekwencjami estetycznymi i poważnymi reperkusjami społecznymi.
Wnioski
Anophthalmia jest niezwykle rzadka. Jego rozpoznanie wymaga badania zarówno etiologii towarzyszącej jak i wad rozwojowych. Jej leczenie jest ciężkie i niedostępne niekiedy dla dzieci z niekorzystnej klasy społecznej, co powoduje uprzedzenia estetyczne. Kwalifikacje szkolenia dla diagnostyki przedporodowej, wczesne zarządzanie przez wykwalifikowanych specjalistów i odpowiedniej platformy technicznej są niezbędne środki w celu zmniejszenia szkód społecznych.
Oświadczenie o etyce
Te przypadki raport przestrzegane doktryny Deklaracji Helsińskiej i został zatwierdzony przez Center Hospital University of Bouaké. Zgodę uzyskano od rodziców noworodka i nie istnieje żaden konflikt interesów dotyczący tego artykułu.
- Speeg-Schatz, Le Marec B. Erreurs génétiques et œil in: Flamment J, Storck D. Œil et pathologie générale Rapport de la soc Fr Ophtalmol. Masson ed, Paris; 1997. P27
- Guthoff R, Klein R, Lieb WE (2004) Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 242: 268-271.
- Mouriaux F, Audo I, Defoort-Dhellemmes S, Labalette P, Guilbert F, et al. (1997) . J Fr Ophtalmol 20: 583-591.
- Roth P, Roth A, Riethmuller D. Ophtalmologie fœtale: l’utilité de l’examen des yeux au cours de l’échographie anténatale. Ophtalmologie 1997; 11: 85-96.
- Romero Caballero MD, López Soler JA, Alcázar Cantos A (2002) . Arch Soc Esp Oftalmol 77: 571-574.
- Busby A, Dolk H, Collin R, Jones RB, Winter R (1998) Compiling a national register of babies born with anophthalmia/microphthalmia in England 1988-94. Arch Dis Child Fetal Neonatal Ed 79: F168-173.
- Araujo J E, Kawanami TE, Nardozza LM, Milani HJ. Oliveira PS, Monron AF (2012) Prenatal diagnosis of bilateral anophthalmia by 3D „reverse face” view ultrasound and magnetic resonance imaging. Taiwan J Obstet Gynecol 51: 616-619.
- Verma AS, Fitzpatrick DR (2007) Anophthalmia and microphthalmia. Orphanet J Rare Dis 2: 47.
- Kouassi FX, Koffi KV, Safede K, Cochard C, Cochener B (2006) . J Fr Ophtalmol 29: e10.
- Diomande I.A, Toure.A, Koffi. K.V, Diomande.G F, Djiguimde.W P, N Habib, A. Ahnoux-zabsonre (2015) Anophthalmia and serious microphthalmia: a summary of the problems associated with antenatal diagnosis and therapeutic refunding in Sub-Saharan. Africa International Medical Case Reports Journal 8: 287-290.
- Stoll C, Dott B, Alembik Y, Roth MP (2012) Associated malformations among infants with anophthalmia and microphthalmia. Birth Defects Res A Clin Mol Teratol 94: 147-152.12.
- Stricker M, Gola R (1990) L’anophtalmie congénitale Chirurgie plastique et réparatrice des paupières et de leurs annexes.Paris : Masson, 177-191.
- Morax.S (1998) Anophtalmies microphtalmies w: Adenis JP, Morax S. Pathologie orbito-palpébrale. Rapport de la Soc Fr Ophtalmol. Paris: Masson. 635-645.
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, et al. (2003) Mutations in SOX2 cause anophthalmia. Nat Genet 33: 461-463.
- Lee B, Song H, Rizzoti K, Son Y, Yoon J, et al. (2013) Genomic code for Sox2 binding uncovers its regulatory role in Six3 activation in the forebrain. Dev Biol 381: 491-501.
- Ye FX, Fan XQ (2012) . Zhonghua Yan Ke Za Zhi 48: 1049-1052.
- Schilter KF, Reis LM, Schneider A, Bardakjian TM, Abdul-Rahman O, et al. (2013) Whole-genome copy number variation analysis in anophthalmia and microphthalmia. Clin Genet 84: 473-481.
- You T, Lv Y, Liu S, Li F, Zhao Y, et al. (2012) Novel OTX2 mutation associated with congenital anophthalmia and microphthalmia in a Han Chinese family. Acta Ophthalmol 90: e501-502.
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, et al. (2004) Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum Mol Genet 13: 315-322.
- Gallardo ME, Rodríguez De Córdoba S, Schneider AS, Dwyer MA, Ayuso C, et al. (2004) Analysis of the developmental SIX6 homeobox gene in patients with anophthalmia/microphthalmia. Am J Med Genet A 129A: 92-94.
- Dufit C, Baggio E, Ruban JM, Buenerd A, Hermier M (1999) Fetal anophthalmos. Problem diagnostyki prenatalnej: opis przypadku. J Fr Ophtalmol 22: 966-969.