En la práctica rutinaria de laboratorio, el diagnóstico del rasgo de beta talasemia suele hacerse por los hallazgos característicos en la evaluación de la hemoglobina y el recuento e índices de glóbulos rojos. En particular, el porcentaje de hemoglobina (Hb) A2 está elevado, mientras que el volumen corpuscular medio (VCM) de los glóbulos rojos y/o la hemoglobina corpuscular media (HCM) están disminuidos. El recuento de glóbulos rojos suele ser normal o estar aumentado, pero puede estar disminuido si el paciente tiene otras causas de anemia.
El rasgo de beta talasemia (también llamado beta talasemia menor o estado de portador de beta talasemia) es un trastorno benigno y heterocigoto que puede distinguirse de los síndromes de beta talasemia más graves (intermedia y mayor) por sus características clínicas y de laboratorio. La beta talasemia intermedia y la mayor se asocian a una mayor gravedad de la anemia, a la dependencia de las transfusiones y a la esplenomegalia, mientras que estas características están ausentes en el rasgo de beta talasemia. En la beta talasemia grave, el nivel de hemoglobina fetal (Hb F) está notablemente aumentado debido a la ausencia de Hb A, y la cantidad de Hb A2 puede no estar aumentada como en el rasgo de beta talasemia. Así que el diagnóstico de laboratorio del rasgo de beta talasemia debería ser relativamente sencillo, ¿correcto?
No siempre. Los dos casos siguientes ilustran situaciones comunes que pueden complicar el diagnóstico.
Caso 1: condiciones concurrentes
La paciente era una mujer afroamericana de 28 años que era positiva para la infección por el VIH-1 y estaba sometida a una terapia antirretroviral altamente activa (HAART) con un régimen de dolutegravir/abacavir/lamivudina. Los resultados de laboratorio se muestran en la Tabla 1.
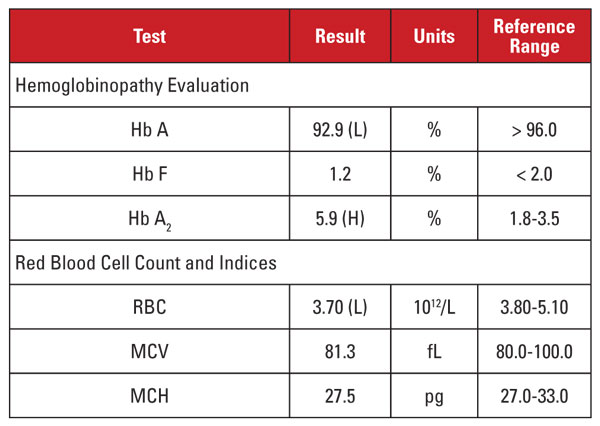
Este caso presentaba una situación interesante. Aunque el nivel de Hb A2 estaba significativamente elevado, el hemograma correspondiente no demostraba microcitosis ni hipocromía. Cuando las características de laboratorio son discordantes para un diagnóstico de rasgo de beta talasemia, es útil considerar otras condiciones que afectan al nivel de Hb A2 y a los índices de glóbulos rojos. En este caso, se encontró una pista importante en la historia clínica del paciente.
Los niveles de Hb A2 tienden a aumentar en condiciones que retrasan la maduración nuclear de los precursores de los glóbulos rojos. Estas condiciones también se asocian con un aumento del VCM.1,2 La causa más común de este fenómeno es la anemia megaloblástica debida a la deficiencia de folato y/o vitamina B12. Sin embargo, varios medicamentos que inhiben la síntesis de ácidos nucleicos tienen un efecto similar, incluyendo la clase de medicamentos contra el VIH llamados inhibidores nucleósidos de la transcriptasa inversa (INTR). La lamivudina en el régimen HAART de este paciente es un medicamento NRTI.
Entonces, ¿debemos concluir que el alto nivel de Hb A2 de este paciente se debió a la medicación contra el VIH, y que el paciente no tenía el rasgo de beta talasemia? No tan rápido. En general, los aumentos de Hb A2 debidos a los NRTI (o a la anemia megaloblástica) son menores que los observados en el rasgo de beta talasemia.1,2 Además, los aumentos tienden a ser proporcionales a los efectos generales del fármaco, que pueden aproximarse a los aumentos del VCM.1,2 En muchos pacientes en tratamiento con NRTI, el VCM es marcadamente alto (a menudo superior a 120 fL), mientras que en nuestro paciente, el VCM estaba hacia el extremo inferior del rango de referencia.
Por lo tanto, no estábamos convencidos de que el nivel muy alto de Hb A2 del paciente se debiera únicamente al tratamiento con lamivudina. Sospechamos que el paciente tenía condiciones concurrentes que aumentaban y disminuían el VCM (es decir, el rasgo de beta talasemia y la terapia con lamivudina, respectivamente), y que ambas contribuían a un nivel elevado de Hb A2. Informamos al médico que nos mandó el caso de que el rasgo de beta talasemia era probable y podía confirmarse mediante un análisis de mutaciones en la beta globina. La posterior secuenciación del gen reveló una mutación heterocigótica de la beta globina asociada a la beta talasemia.
Caso 2: factor étnico
La paciente era una mujer vietnamita embarazada de 35 años a la que se le estaba realizando un cribado de hemoglobinopatía. Los resultados de laboratorio se muestran en la tabla 2.
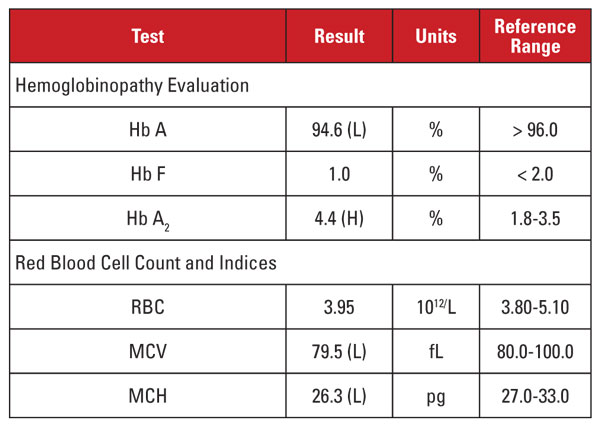
De nuevo, el nivel elevado de Hb A2 era discordante con microcitosis e hipocromía mínimas o ausentes. En este caso, pudimos descartar factores de confusión comunes como la deficiencia de folato, vitamina B12 o hierro, la disfunción tiroidea o los efectos de la medicación. Basándonos en el origen étnico de la paciente, sospechamos otra causa importante de discordancia: el rasgo de beta talasemia coherente con la alfa talasemia menor. Cuando un paciente hereda ambas afecciones, el VCM y la HCM suelen normalizarse porque las cadenas de globina alfa y beta están presentes en cantidades relativamente equilibradas en los glóbulos rojos en desarrollo.2
Recomendamos la realización de análisis genéticos para comprobar la existencia de mutaciones de la talasemia alfa y beta, con el fin de evaluar plenamente el riesgo para la madre embarazada y el feto. La posterior secuenciación de la beta globina reveló una mutación beta plus talasémica heterocigótica, mientras que el análisis de las deleciones de la alfa globina reveló una deleción heterocigótica de dos genes del sudeste asiático (SEA), consistente con la -/α alfa talasemia menor. Sobre la base de estos resultados, se indicó el análisis genético del padre para evaluar el riesgo de que el feto heredara un síndrome de talasemia clínicamente grave.
Talasemias alfa y beta
Las talasemias se encuentran entre los trastornos hereditarios más comunes de la humanidad. Son muy frecuentes donde la malaria ha sido endémica, y ahora son comunes en todas las partes del mundo debido a la migración de las poblaciones humanas. Las talasemias están causadas por mutaciones que reducen la expresión del gen de la globina en los precursores de los glóbulos rojos. Los trastornos se clasifican en función de los genes de globina mutados (por ejemplo, alfa frente a beta) y de la gravedad de la enfermedad, que está relacionada con el hecho de que las mutaciones se hereden de forma heterocigota u homocigota/ heterocigota compuesta. La principal forma de hemoglobina en adultos es la Hb A, un tetrámero de dos cadenas de globina alfa y dos beta. En la talasemia alfa, la expresión de la globina alfa es deficiente y hay un exceso correspondiente de cadenas de globina beta.
Este patrón se invierte en la talasemia beta. En el contexto de la deficiencia y el desequilibrio de las cadenas de globina, el contenido de hemoglobina de los glóbulos rojos disminuye, lo que provoca microcitosis e hipocromía. Además, el exceso de cadenas de globina alfa o beta forma tetrámeros inestables que causan hemólisis. Las talasemias alfa y beta se distinguen por la cantidad de la hemoglobina adulta menor Hb A2, un tetrámero de dos cadenas de globina alfa y dos delta. La Hb A2 está aumentada en la beta talasemia porque la falta relativa de beta globina permite que se incorporen más cadenas delta a la hemoglobina.
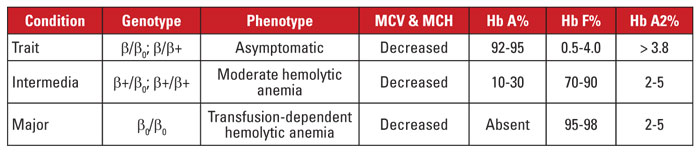
La beta talasemia está causada por mutaciones en el locus del gen de la beta globina en el cromosoma 11.3,4 La mayoría de las mutaciones son pequeñas sustituciones, inserciones o deleciones de nucleótidos, aunque en casos raros se identifican grandes deleciones. Dependiendo de la mutación, la expresión de la beta globina se reduce parcialmente (talasemia beta plus) o completamente (talasemia beta cero). El rasgo de beta talasemia está causado por una mutación heterocigótica. Esta condición es asintomática y se caracteriza por un aumento de la Hb A2, microcitosis de los glóbulos rojos y ninguna anemia hemolítica significativa. En cambio, la beta talasemia mayor (anemia de Cooley) está causada por mutaciones beta-cero homocigóticas. La evaluación de la hemoglobina revela un predominio de la Hb F, ausencia de la Hb A y una Hb A2 normal o aumentada. La beta talasemia mayor se caracteriza por una anemia hemolítica grave, dependiente de las transfusiones, con la consiguiente esplenomegalia y deformidades óseas. Las transfusiones recurrentes provocan una sobrecarga de hierro, que es la principal causa de morbilidad y mortalidad. La beta talasemia intermedia es clínicamente heterogénea, con la mayoría de los síntomas relacionados con la anemia hemolítica moderada. La dependencia de la transfusión es infrecuente porque la expresión de la beta globina no está ausente. Debido al gran número de mutaciones asociadas a la beta talasemia, el diagnóstico genético suele requerir la secuenciación del gen. Los síndromes de beta talasemia se resumen en la Tabla 3.
La beta talasemia está causada por mutaciones en el locus del gen de la alfa globina en el cromosoma 16.5 Los genes de la alfa globina están duplicados en el cromosoma 16, por lo que un individuo normal tiene cuatro copias. Las mutaciones más comunes son grandes deleciones que afectan a uno o ambos genes del locus. Se ha descrito un número bastante limitado de deleciones, con prevalencia en diferentes poblaciones; por ejemplo, las mutaciones grandes que eliminan ambos genes alfa son muy comunes en personas de la etnia del sudeste asiático. Estas mutaciones también son frecuentes en las poblaciones mediterráneas, pero son raras en las personas de etnia africana. En cambio, las supresiones de genes alfa únicos tienen una distribución generalizada. La pérdida de uno de los cuatro genes de globina alfa se conoce como estado de portador silencioso.
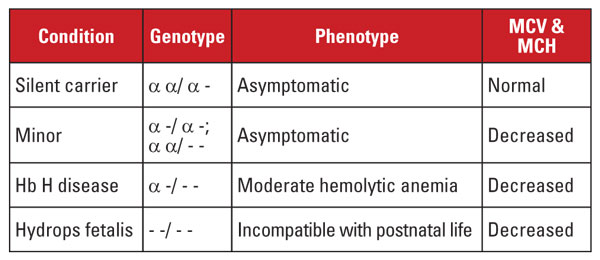
Esta condición es asintomática y suele estar asociada a índices normales de glóbulos rojos. La pérdida de dos genes de globina alfa se denomina alfa talasemia menor. Esta condición puede ocurrir debido a deleciones homocigóticas de un gen o a deleciones heterocigóticas de dos genes. El recuento de glóbulos rojos y los índices son indistinguibles del rasgo de beta talasemia, pero el nivel de Hb A2 es normal. La pérdida de tres genes alfa se denomina enfermedad de Hb H. (La Hb H es un tetrámero de cadenas de beta globina.) La enfermedad de la Hb H es típicamente una anemia hemolítica moderada asociada con esplenomegalia y cambios óseos. La pérdida de los cuatro genes alfa se denomina hidropesía fetal Hb Barts (la Hb Barts es un tetrámero de cadenas de globina gamma fetal). Esta condición es incompatible con la vida postnatal. La Hb H y la Hb Barts pueden identificarse en las evaluaciones de hemoglobina y desempeñan un papel en el diagnóstico postnatal de la alfa talasemia.5
El diagnóstico genético suele realizarse mediante ensayos de reacción en cadena de la polimerasa (PCR) que se dirigen a las deleciones comunes. La secuenciación del gen de la alfa globina también está disponible para detectar mutaciones raras. Los síndromes de alfa talasemia se resumen en la Tabla 4.
Cuando se necesitan pruebas genéticas
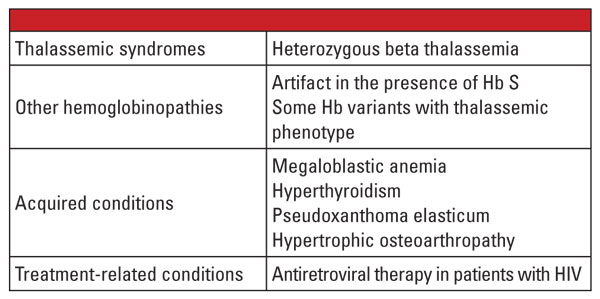
En conclusión, las formas heterocigotas de alfa y beta talasemia son muy comunes y se identifican con frecuencia en el laboratorio clínico. La evaluación de la hemoglobina y otros datos clínicos y de laboratorio rutinarios suelen ser suficientes para el diagnóstico. Las pruebas genéticas no son necesarias en la mayoría de los casos, pero pueden ser muy importantes en el contexto de la planificación familiar. Si se identifican mutaciones de talasemia en ambos padres, puede haber un riesgo significativo para el feto de heredar una talasemia grave. En el caso de la hidropesía fetal, es probable la muerte del feto y existe un riesgo significativo para la madre de sufrir complicaciones en el embarazo.5 Por lo tanto, es importante ser consciente de las variables que complican el diagnóstico de laboratorio de las talasemias y reconocer cuándo está indicado realizar pruebas genéticas confirmatorias. Por ejemplo, como se muestra en el primer informe de caso, el diagnóstico del rasgo de beta talasemia puede ser un reto si hay condiciones coexistentes que afectan al nivel de Hb A2. Los factores que se sabe que aumentan la Hb A2 se enumeran en la Tabla 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biología, relevancia clínica y un posible objetivo para mejorar la enfermedad de células falciformes. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Revisión de pruebas genéticas. Alfa-talasemia. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH recommendations for the measurement of haemoglobin A2. Int J Lab Hematol. 2012;34(1):1-13.