Abstrakt
Úvod: Kongenitální anoftalmie je klinická absence oka při narození. Je důsledkem nedostatečného vývoje nebo regrese primárního zrakového váčku během embryonálního života. Vyskytuje se vzácně a může být izolovaná nebo spojená s jinými očními nebo celkovými vrozenými vadami.
Pozorování: Uvádíme případy tří dětí z jedné rodiny ve věku od 4 měsíců do 8 let s oboustrannou vrozenou anoftalmií. Jejich anamnéza se vyznačuje nedostatkem konzultací a prenatálních laboratorních vyšetření. Oftalmologické vyšetření objektivizovalo, při otevření víček oběma očima, výplň dutiny spojivkovou tkání ve 3 případech nasvědčující oboustranné anoftalmii, potvrdil oční ultrazvuk.
Diskuse: Kongenitální anoftalmie je vzácná malformace. Může být izolovaná nebo integrovaná do malformačního syndromu. Klinický obraz je většinou jednostranný. Diagnostika je převážně klinická a potvrzuje se pomocí očního ultrazvuku. Příčiny jsou různé, zastoupeny jsou chromozomální aberace, genetické mutace, otravy a infekce získané během těhotenství.
Závěr: Zjištění vrozené anoftalmie vyžaduje komplexní přezkoumání za účelem hledání etiologie. Důležitým bodem zůstává také psychologická podpora rodičů.
Klíčová slova
anoftalmie, vrozená, oční ultrazvuk, chromozomální aberace
Úvod
Anoftalmie je klinické chybění oka. Jedná se o extrémně vzácnou oční abnormalitu. Existuje mnoho etiologií, například chromozomální aberace, genetické mutace, intoxikace a infekce. Vrozená anoftalmie může být izolovaná nebo spojená se závažnými polymorformními syndromy. Způsobuje na jedné straně mikroorbitální a homolaterální kraniofaciální hemiatrofii a na druhé straně špatný vývoj přídatných orgánů, což představuje problémy při protetické adaptaci. Diagnostika se zlepšila díky prenatálnímu zobrazování a terapeutické řešení v podstatě zahrnuje orbitální protézy. Uvádíme 3 případy oboustranné kongenitální anoftalmie v jedné rodině.
Případ 1
Jedná se o chlapce ve věku 8 let, narozeného v termínu, přijatého ke konzultaci pro oboustranný pokles zrakové ostrosti. Biologické a orbitocerebrální prenatální CT-vyhodnocení nebylo možné. Klinické oftalmologické vyšetření odhalilo absenci světelného vjemu na obou očích; oboustrannou anoftalmii; apendixy jsou přítomny a normální a při otevření víček spojivková struktura vyplňující celou očnicovou dutinu bez oční bulvy. Vyžádané CT vyšetření očnice a mozku potvrdilo absenci oční koule s přítomností zbytkového tuku, okohybných svalů a atrofií kalibru zrakového nervu v očnicové dutině (obrázek 1). Celková bilance byla normální. A magnetickou rezonanci nebylo možné realizovat vzhledem k finančním potížím rodičů, stejně jako genetickou rozvahu (karyotypizaci). Byla navržena chirurgická léčba s umístěním protézy, která však nebyla realizována pro nedostatek odpovídající technické platformy. (Obrázek 2)
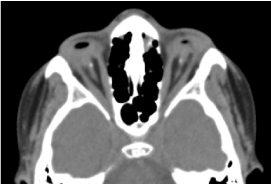
Obrázek 1. CT vyšetření mozku očnice: absence oční koule s reziduálním tukem, svaly a atrofií kalibru otocerválního nervu v dutině očnice.

Obrázek 2: Oční koule. 2. Osmileté dítě vykazující oboustranný anoftalmus při otevření víček
Případ 2
Jednalo se o osmileté dítě mužského pohlaví, u kterého nebylo provedeno prenatální vyšetření (biologické a ultrazvukové echografické). Byl přijat do oftalmologické poradny pro oboustranný pokles zrakové ostrosti. Klinickým oftalmologickým vyšetřením byla zjištěna absence světelného vjemu na obou očích; oboustranná anoftalmie, apendixy bez zvláštností a při otevření víček spojivková struktura vyplňující dutinu). vyžádané orbitocerebrální CT vyšetření potvrdilo absenci oční koule s přítomností reziduálního tuku, okohybných svalů a atrofii kalibru zrakového nervu v dutině očnice (obr. 3). Celkové vyšetření tohoto dítěte bylo normální. A magnetická rezonance ani karyotyp nemohly být provedeny ze stejných důvodů, které byly zmíněny dříve, stejně jako lékařský management. (Obrázek 4)
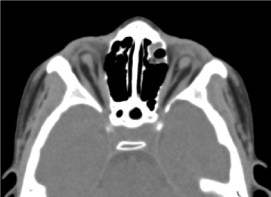
Obrázek.3. absence oční koule s reziduálním tukem, svaly a atrofií kalibru otocerválního nervu v očnicové dutině.
Případ 3
Jedná se o 4měsíčního kojence ženského pohlaví, narozeného v termínu a s dobrým psychomotorickým vývojem a staturou, přijatého do oftalmologické poradny pro oboustranný pokles zrakové ostrosti a trvalý zákal očních víček od narození. V anamnéze kojence jsou zaznamenány dvě prenatální konzultace, ale žádné prenatální biologické a ultrazvukové vyšetření. Klinickým oftalmologickým vyšetřením byla zjištěna absence světelného stopu; oboustranná anoftalmie; normální aspekty příklopek a k otevírání víček, prázdná oční koule dávající mikroftalmický vzhled, detaily duhovky nejsou analyzovatelné a absence přední komory. Pediatrická konzultace při pátrání po jiných malformacích se vrátila bez konkrétních údajů na klinické úrovni.
Vyžádaná orbitocerebrální tomografie potvrdila absenci oční koule s přítomností reziduálního tuku, okohybných svalů a atrofii kalibru zrakového nervu v očnicové dutině (obrázek 5). Magnetickou rezonanci a karyotypizaci nebylo možné provést; stejně jako jeho medikamentózní léčbu (obr. 6).

Obrázek 4. Šestileté dítě s oboustrannou anoftalmií při otevření víček.
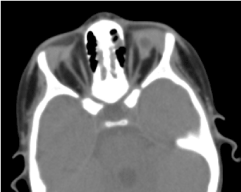
Obrázek 5. Dítě s anoftalmií při otevření víček. 5. Absence oční koule se zbytkem vezikuly, svalů a atrofií otocerválního nervu v orbitální dutině.

Obr.6.. 4měsíční dítě; při otevření víček jsme si mohli všimnout, zejména u pravého oka, nákresu rohovky, ale duhovka není individualizovatelná.
Diskuse
Vrozená anoftalmie neboli klinická absence oka při narození je vzácná malformace . Její výskyt je variabilní. U Mouriauxe a Rotha se odhaduje na méně než jeden případ na 10000 porodů . Podle španělského průzkumu by to bylo asi 21,34 na 100 000 porodů a podle britské studie 10 na 100 000 porodů . U Aranjo , je její výskyt 0,6 na 10000 živě narozených dětí. Tento rozdíl v incidenci nachází vysvětlení ve variabilitě výskytu této vady. Četnost anoftalmie není ovlivněna rasou ani pohlavím. Anoftalmie může být izolovaná nebo integrovaná do polymalformačního syndromu v jedné třetině případů . Toto pozorování je v souladu s pozorováními několika autorů. U Verna se tedy anoftalmie i mikroftalmie mohou vyskytovat izolovaně nebo mohou být integrovány do Patauova syndromu (trizomie13) nebo jiných syndromů přibližně v jedné třetině případů. Kouassi rovněž popsal případ oboustranné kongenitální anoftalmie při Patauově syndromu. Stejně tak Diomandé v Bouaké, který popsal případ skutečné kongenitální anoftalmie spojené s těžkou mikroftalmií u novorozence bez dalších pozorovaných klinických abnormalit.
Stoll navíc poznamenal, že 90 % anoftalmií mělo přidružené malformace. Proto je v přítomnosti jakékoli vrozené anoftalmie nebo extrémní mikroftalmie důležité provést důkladné pediatrické vyšetření s cílem vyhledat přidružené abnormality, někdy závažné, jako je anencefalie nebo ageneze celého optického traktu . Klinická anoftalmie je zřídka oboustranná. Moderní paraklinická vyšetření, jako je počítačová tomografie a magnetická rezonance, umožňují vyšetřit související malformace. V našich pozorováních nebylo možné tyto bilance realizovat vzhledem k socioekonomickým potížím rodičů.
Podle Speega – Schatze , existují tři typy vrozené anoftalmie: primární anoftalmie, sekundární anoftalmus a konsekutivní sekundární anoftalmie jako důsledek absence invaginace optické jamky, úplného zastavení vývoje přední neurální trubice nebo degenerace optického váčku po jeho invaginaci. Podle Strickera by se vrozená anoftalmie mohla analyzovat a klasifikovat podle dvou způsobů.
Z anatomického hlediska by bylo nutné odlišit pravou anoftalmii od embryopatií charakterizovaných absencí jakýchkoli očních obrysů. To by potvrdilo pouze histologické vyšetření.
Z klinického hlediska je třeba oddělit klinickou anoftalmii neboli extrémní mikroftalmii od jiných, mírnějších mikroftalmií s někdy lepší funkční prognózou. V pozdějších případech je více nebo rudimentární oční bulva (jako v našich 3. klinických případech) někdy doprovázená kolobomatózní cystou schopnou fungovat jako fyziologická expanzivní protéza. Klinicky není přítomna žádná oční struktura, ale oční ultrazvuk, počítačová tomografie a magnetická rezonance by ukázaly přítomnost náčrtu očního bulbu a zrakového nervu.
Morax , však poznamenal, že pravá anoftalmie neexistuje. Podle něj by v nejvýraznějších klinických případech neurozobrazení ukázalo přítomnost zrakového nervu a drobného náčrtu oka. V našich prvních dvou klinických případech oční ultrazvuk prokázal nepřítomnost oční nerovnosti. Vzhledem k absenci výsledků CT vyšetření a magnetické rezonance však nemůžeme formálně uzavřít, že není přítomen oční nerv a žádná oční rýha. etiologie anoftalmie je složitá a různorodá. Může být dědičná na základě chromozomálních aberací, zejména trizomie 13 nebo trizomie 18. V tomto případě se jedná o anoftalmii s mozkovými malformacemi nebo spojenou s dysplazií střední čáry . Další genetickou etiologii představují chromozomální mutace, které se mohou vyskytnout při chromozomálních delecích nebo nezávisle na jakékoli aberaci karyotypu. Jedná se především o anomálie SOX2. Fantes diagnostikoval mikrodeleci spojenou s translokací t (3; 11) (q26-3 p11-2) a mutací genu SOX2 u pacienta s oboustrannou anoftalmií. Předpokládá se, že haploinsuficience genu SOX2 je zodpovědná za anoftalmii, mikroftalmii, septo-optickou dysplazii a defekt ve vývoji předního mozku . Ye poznamenal, že defekt SOX2 by mohl způsobit mnohočetnou poruchu organického systému včetně anoftalmie a mikroftalmie. Na vzniku anoftalmie by se podílely i další geny, například PAX6, OTX2, CHV10, RAX2, SIX6, NF1, TWIST, SHH, BMP4 a BMP7. Podle Schiltera by anoftalmii vysvětlovalo méně než 40 % známých genetických mutací. Za anoftalmii nebo mikroftalmii by podle něj byla zodpovědná mutace genů NF1, PAX6, SOX2. Popsal jste mutaci genu OTX2 spojenou s anoftalmií a kongenitální mikroftalmií v čínské rodině. Veronika pozorovala u jednoho pacienta mutaci genu RAX2 zodpovědnou za jednostrannou anoftalmii. Mutace v genu TWIST při deleci chromozomu 7 mohou vést k dysgenezi obličeje s anoftalmií . Identifikaci mutace alely genu SIX6 u pacienta s oboustrannou anoftalmií zaznamenal Gallardo .
Bez karyotypizace nejsme schopni tyto dědičné anoftalmy v naší studii přiřadit ke konkrétnímu typu mutace. Navíc Patauův syndrom, který je jednou z hlavních etiologií, nelze u našich pacientů klinicky zachytit.
Z těchto všech etiologických faktorů se zdá, že teorie genetické mutace odpovídá našim klinickým případům, skutečně tyto 3 případy pocházejí ze stejné rodiny, ale také z konsanguinního spojení, protože sami pocházejí ze stejné rodiny, protože jsou bratranci a sestřenice. Je však třeba poznamenat, že nedostatek vitaminu A, vystavení rentgenovému záření, zneužívání rozpouštědel, užívání thalidominu a infekce získané během těhotenství by byly také faktory prostředí . Diagnózu anoftalmie lze stanovit pre- nebo postnatálně na základě kombinace klinických příznaků, zobrazovacích metod a genetické analýzy. Dufit , který použil nové zobrazovací techniky (fetální MRI), popsal případ fetální anoftalmie spojené s mnohočetnými malformacemi, které vedly k ukončení těhotenství.
U dětí se doporučuje protetická léčba pomocí zvětšujících se konformátorů, nebo dokonce techniky rozšíření očnice pomocí nafukovacích protéz, a to do 2 let věku. U dospělých se však tento typ léčby stává zbytečným a nedoporučuje se. Terapeutický režim musí zahrnovat zvětšení očnice různými osteotomiemi spojenými současně nebo sekundárně s obnovou spojivkového vaku slizničním štěpem nebo dermoepidermálním štěpem. Poté dojde k obnově víček rekonstrukcí, repozicí a korekcí malpozice na protéze . V našich rozvojových zemích je tento typ těžké a někdy neuspokojivé léčby obtížně proveditelný někdy neškolenými týmy. Navíc míra chudoby rodičů omezuje jejich dostupnost léčby, což má estetické důsledky a velký sociální dopad.
Závěr
Anoftalmie je extrémně vzácná. Její diagnostika zahrnuje vyšetření přidružených etiologií i malformací. Její léčba je těžká a někdy nedostupná pro děti z nepříznivé sociální vrstvy, což vede k estetickým předsudkům. Kvalifikační příprava pro prenatální diagnostiku, včasná léčba vyškolenými odborníky a odpovídající technická platforma jsou základními prostředky ke snížení společenské škodlivosti.
Etické prohlášení
Tato zpráva o případech se řídila zásadami Helsinské deklarace a byla schválena Centrem nemocnice University of Bouaké. Od rodičů novorozence byl získán souhlas a tento článek není ve střetu zájmů.
- Speeg-Schatz, Le Marec B. Erreurs génétiques et œil in: Flamment J, Storck D. Œil et pathologie générale Rapport de la soc Fr Ophtalmol. Masson ed, Paris; 1997. P27
- Guthoff R, Klein R, Lieb WE (2004) Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 242: 268-271.
- Mouriaux F, Audo I, Defoort-Dhellemmes S, Labalette P, Guilbert F a další (1997) . J Fr Ophtalmol 20: 583-591.
- Roth P, Roth A, Riethmuller D. Ophtalmologie fœtale: l’utilité de l’examen des yeux au cours de l’échographie anténatale. Ophtalmologie 1997; 11: 85-96.
- Romero Caballero MD, López Soler JA, Alcázar Cantos A (2002) . Arch Soc Esp Oftalmol 77: 571-574.
- Busby A, Dolk H, Collin R, Jones RB, Winter R (1998) Sestavení národního registru dětí narozených s anoftalmií/mikroftalmií v Anglii v letech 1988-1994. Arch Dis Child Fetal Neonatal Ed 79: F168-173.
- Araujo J E, Kawanami TE, Nardozza LM, Milani HJ. Oliveira PS, Monron AF (2012) Prenatální diagnostika bilaterální anoftalmie pomocí 3D ultrazvuku a magnetické rezonance v zobrazení „reverse face“. Taiwan J Obstet Gynecol 51: 616-619.
- Verma AS, Fitzpatrick DR (2007) Anoftalmie a mikroftalmie. Orphanet J Rare Dis 2: 47.
- Kouassi FX, Koffi KV, Safede K, Cochard C, Cochener B (2006) . J Fr Ophtalmol 29: e10.
- Diomande I.A, Toure.A, Koffi. K.V, Diomande.G F, Djiguimde.W P, N Habib, A. Ahnoux-zabsonre (2015) Anoftalmie a závažná mikroftalmie: shrnutí problémů spojených s prenatální diagnostikou a terapeutickou refundací v subsaharských zemích. Africa International Medical Case Reports Journal 8: 287-290.
- Stoll C, Dott B, Alembik Y, Roth MP (2012) Associated malformations among infants with anophthalmia and microphthalmia. Birth Defects Res A Clin Mol Teratol 94: 147-152.12.
- Stricker M, Gola R (1990) L’anophtalmie congénitale Chirurgie plastique et réparatrice des paupières et de leurs annexes. paříž : Masson, 177-191.
- Morax.S (1998) Anophtalmies microphtalmies in: Adenis JP, Morax S. Pathologie orbito-palpébrale. Rapport de la Soc Fr Ophtalmol. Paris: Masson. 635-645.
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, et al. (2003) Mutace v SOX2 způsobují anoftalmii. Nat Genet 33: 461-463.
- Lee B, Song H, Rizzoti K, Son Y, Yoon J, et al. (2013) Genomický kód pro vazbu Sox2 odhaluje jeho regulační roli při aktivaci Six3 v předním mozku. Dev Biol 381: 491-501.
- Ye FX, Fan XQ (2012) . Zhonghua Yan Ke Za Zhi 48: 1049-1052.
- Schilter KF, Reis LM, Schneider A, Bardakjian TM, Abdul-Rahman O, et al. (2013) Whole-genome copy number variation analysis in anophthalmia and microphthalmia. Clin Genet 84: 473-481.
- You T, Lv Y, Liu S, Li F, Zhao Y, et al. (2012) Novel OTX2 mutation associated with congenital anophthalmia and microphthalmia in a Han Chinese family. Acta Ophthalmol 90: e501-502.
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, et al. (2004) Mutace v lidském homeoboxovém genu RAX u pacienta s anoftalmií a sklerokorneou. Hum Mol Genet 13: 315-322.
- Gallardo ME, Rodríguez De Córdoba S, Schneider AS, Dwyer MA, Ayuso C, et al. (2004) Analysis of the developmental SIX6 homeobox gene in patients with anophthalmia/microphthalmia. Am J Med Genet A 129A: 92-94.
- Dufit C, Baggio E, Ruban JM, Buenerd A, Hermier M (1999) Fetální anoftalmus. Problém prenatální diagnostiky: kazuistika. J Fr Ophtalmol 22: 966-969.