Abstract
Introducere: Anoftalmia congenitală reprezintă absența clinică a ochiului la naștere. Ea rezultă din lipsa de dezvoltare sau regresia veziculei optice primare în timpul vieții embrionare. Este rară și poate fi izolată sau asociată cu alte malformații oculare sau generale la naștere.
Observații: Raportăm cazurile a trei copii din aceeași familie, cu vârste cuprinse între 4 luni și 8 ani, cu anoftalmie congenitală bilaterală. Istoricul lor este marcat de lipsa consultului și a testelor de laborator prenatale. Examenul oftalmologic a obiectivat, prin deschiderea pleoapelor cu doi ochi, o umplere a cavității de către țesutul conjunctival în 3 cazuri, sugestivă pentru anoftalmie bilaterală, confirmată de ecografia oculară.
Discuție: Anoftalmia congenitală este o malformație rară. Ea poate fi izolată sau integrată unui sindrom malformativ. Tabloul clinic este de cele mai multe ori unilateral. Diagnosticul este în principal clinic și confirmat prin ecografie oculară. Cauzele sunt variate, reprezentate de aberații cromozomiale, mutații genetice, intoxicații și infecții dobândite în timpul sarcinii.
Concluzie: Descoperirea unei anoftalmii congenitale impune un bilanț complet în vederea căutării etiologiei. Sprijinul psihologic al părinților rămâne, de asemenea, un obiectiv important.
Cuvinte cheie
anoftalmie, congenitală, ecografie oculară, aberații cromozomiale
Introducere
Anoftalmia este lipsa clinică a ochiului. Este o anomalie oculară extrem de rară. Există numeroase etiologii, cum ar fi aberațiile cromozomiale, mutațiile genetice, intoxicațiile și infecțiile. Anaftalmia congenitală poate fi izolată sau asociată cu sindroame polimorformative severe. Ea este responsabilă, pe de o parte, de o micro-orbită și de o hemi-atrofie cranio-facială homolaterală și, pe de altă parte, de o slabă dezvoltare a apendicelor care pune probleme în timpul adaptării protetice. Diagnosticul a fost îmbunătățit prin imagistică prenatală, iar managementul terapeutic implică în mod esențial proteze orbitale. Raportăm 3 cazuri de anoftalmie congenitală bilaterală în aceeași familie.
Cazul 1
Este vorba de un copil de sex masculin în vârstă de 8 ani, născut la termen, primit în consultație pentru o scădere bilaterală a acuității vizuale. Evaluarea prenatală biologică și CT-scan orbito-cerebrală nu a fost posibilă. Examenul clinic oftalmologic a evidențiat absența percepției luminoase la ambii ochi; Anoftalmie bilaterală; apendicele sunt prezente și normale, iar la deschiderea pleoapelor, o structură conjunctivală care umple întreaga cavitate orbitală fără globul ocular. Tomografia computerizată orbito-cerebrală solicitată a confirmat absența globului ocular cu prezența grăsimii reziduale, a mușchilor oculomotori și o atrofie de calibru a nervului optic în cavitatea orbitală (figura 1). Bilanțul general a fost normal. Iar imagistica prin rezonanță magnetică nu a putut fi realizată având în vedere dificultățile financiare ale părinților, precum și bilanțul genetic (cariotiparea). Tratamentul chirurgical cu plasarea unei proteze a fost propus, dar nu s-a concretizat din cauza lipsei unei platforme tehnice adecvate. (Figura 2)
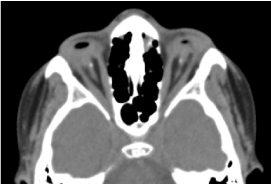
Figura 1. CT-scanare cerebrală a orbitei: absența globului ocular cu grăsime reziduală, mușchi și o atrofie a calibrului nervului otocervical în cavitatea orbitală.

Figură. 2. Copil în vârstă de 8 ani care prezintă anoftalmie bilaterală la deschiderea pleoapelor
Cazul 2
Este vorba de un copil de sex masculin în vârstă de 8 ani, la care nu s-a efectuat o evaluare prenatală (biologică și ecografică). A fost primit în consult oftalmologic pentru o scădere bilaterală a acuității vizuale. Examenul clinic oftalmologic a constatat absența percepției luminoase la ambii ochi; O anoftalmie bilaterală, anexe fără particularități și la deschiderea pleoapelor, o structură conjunctivală care umplea cavitatea). tomografia computerizată orbito-cerebrală solicitată a confirmat absența globului ocular cu prezența grăsimii reziduale, a mușchilor oculomotori și o atrofie de calibru a nervului optic în cavitatea orbitală (figura 3). Examenul general al acestui copil a fost normal. Iar imagistica prin rezonanță magnetică, precum și cariotipul nu au putut fi efectuate din aceleași motive menționate anterior, la fel ca și managementul medical. (Figura 4)
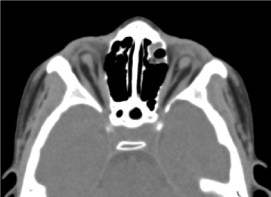
Figura.3. absență a globului ocular cu grăsime reziduală, mușchi și o atrofie a calibrului nervului otocervical în cavitatea orbitală.
Cazul 3
Este cel al unui sugar de sex feminin în vârstă de 4 luni, născut la termen și având o bună dezvoltare psihomotorie și staturo-ponderală, primit în consult oftalmologic pentru scăderea bilaterală a acuității vizuale și ocluzia permanentă a pleoapelor de la naștere. Anamneza sugarului este marcată de două consultații prenatale, dar fără analize biologice și ecografice prenatale. Examenul clinic oftalmologic a constatat absența urmăririi luminoase; Anoftalmie bilaterală; Aspecte normale ale apendicelor și la deschiderea pleoapelor, un glob ocular gol care dă un aspect microftalmic, detaliile irisului nu sunt analizabile și absența unei camere anterioare. Consultația pediatrică în căutarea altor malformații s-a întors fără particularități pe plan clinic.
Tomografia orbito-cerebrală solicitată a confirmat absența globului ocular cu prezența grăsimii reziduale, a mușchilor oculomotori și o atrofie de calibru a nervului optic în cavitatea orbitală (figura 5). Imagistica prin rezonanță magnetică și cariotipul nu au putut fi efectuate; la fel ca și managementul său medical (Figura 6).

Figura.4. Copil de 6 ani cu anoftalmie bilaterală la deschiderea pleoapelor.
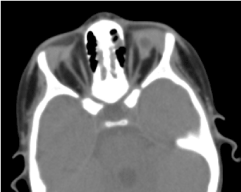
Figura.5. Copil de 6 ani cu anoftalmie bilaterală la deschiderea pleoapelor. 5. O absență a globului ocular cu o veziculă reziduală, mușchi și o atrofie a nervului otocervical în cavitatea orbitală.

Figura.6. copil de 4 luni; la deschiderea pleoapelor s-a putut observa, mai ales la ochiul drept o schiță de cornee dar irisul nu este individualizabil.
Discuție
Anoftalmia congenitală sau absența clinică a ochiului la naștere este o malformație rară . Incidența sa este variabilă. Este estimată la mai puțin de un caz la 10000 de nașteri pentru Mouriaux și Roth . Ar fi de aproximativ 21,34 la 100.000 de nașteri conform unui studiu spaniol și de 10 la 100.000 de nașteri conform unui studiu britanic . Pentru Aranjo , incidența sa este de 0,6 la 10000 de copii născuți vii. Această diferență de incidență își găsește explicația în variabilitatea de apariție a acestei malformații. Frecvența anoftalmiei nu este influențată de rasă sau de sex. Anoftalmia poate fi izolată sau integrată într-un sindrom polimalformativ în o treime din cazuri . Această observație este în conformitate cu cele ale mai multor autori. Astfel, pentru Verna , atât anoftalmia cât și microftalmia pot apărea izolat sau pot fi integrate într-un sindrom Patau (trisomia13) sau în alte sindroame în aproximativ o treime din cazuri. Kouassi a raportat, de asemenea, un caz de anoftalmie congenitală bilaterală în cadrul sindromului Patau. La fel și Diomandé din Bouaké, care a raportat un caz de anoftalmie congenitală reală asociată cu microftalmie severă la un nou-născut fără alte anomalii clinice observate.
În plus, Stoll a observat că 90% dintre anoftalmii au malformații asociate. Prin urmare, în prezența oricărei anoftalmii congenitale sau microftalmii extreme, este important să se efectueze o evaluare pediatrică amănunțită în căutarea unor anomalii asociate, uneori severe, cum ar fi anencefalia sau agenezia întregului tract optic . Anoftalmia clinică este rareori bilaterală. Investigațiile paraclinice moderne, cum ar fi tomografia computerizată și imagistica prin rezonanță magnetică, fac posibilă investigarea malformațiilor asociate. În observațiile noastre, aceste bilanțuri nu au putut fi realizate având în vedere dificultățile socio-economice ale părinților.
Potrivit lui Speeg – Schatz , există trei tipuri de anoftalmie congenitală: anoftalmie primară, anoftalmie secundară și anoftalmie secundară consecutivă ca urmare a absenței invaginării fosei optice, a încetării complete a dezvoltării tubului neural anterior sau a degenerării veziculei optice după invaginarea acesteia. Pentru Stricker , anoftalmia congenitală ar putea fi analizată și clasificată după două modalități.
Din punct de vedere anatomic, ar fi necesar să se distingă anoftalmia adevărată de embriopatiile caracterizate prin absența oricărui contur ocular. Numai examenul histologic ar putea confirma acest lucru.
Din punct de vedere clinic, anoftalmia clinică sau microftalmia extremă trebuie separată de alte microftalmii mai moderate, cu prognostic funcțional uneori mai bun. În cazurile mai târzii, există un glob ocular mai mult sau rudimentar (ca în al 3-lea caz clinic al nostru) însoțit uneori de un chist colobomatos capabil să acționeze ca o proteză expansivă fiziologică. Din punct de vedere clinic, nu există nici o structură oculară, dar o ecografie oculară, o tomografie computerizată și o imagistică prin rezonanță magnetică ar arăta prezența unei schițe a globului ocular și a nervului optic.
Cu toate acestea, Morax , a remarcat că nu există o adevărată anoftalmie. Potrivit acestuia, în cele mai marcate cazuri clinice, neuroimagistica ar arăta prezența unui nerv optic și a unei mici schițe oculare. În primele noastre două cazuri clinice, ecografia oculară a arătat o absență a schiței oculare. Cu toate acestea, în absența rezultatelor CT-scan și IRM, nu putem concluziona în mod formal că nu există nerv optic ocular și nici nerv optic. etiologia anoftalmiei este complexă și diversă. Ea poate fi ereditară prin aberații cromozomiale, în special trisomia 13 sau trisomia 18. În acest caz, este vorba de anoftalmie cu malformații cerebrale sau asociată cu o displazie a liniei mediane . Alte etiologii genetice sunt reprezentate de mutații cromozomiale care pot apărea în cursul unor deleții cromozomiale sau independent de orice aberație a cariotipului. Anomaliile SOX2 sunt cele principale. Fantes a diagnosticat o microdeleție asociată cu o translocație t (3; 11) (q26-3 p11-2) și o mutație a genei SOX2 la un pacient cu anoftalmie bilaterală. Se crede că haploinsuficiența genei SOX2 este responsabilă de anoftalmie, microftalmie, displazie septo-optică și un defect de dezvoltare a creierului anterior . Ye a observat că defectul SOX2 ar cauza o tulburare multiplă a sistemului organic, inclusiv anoftalmie și microftalmie. Alte gene, cum ar fi PAX6, OTX2, CHV10, RAX2, SIX6, NF1, TWIST, SHH, BMP4 și BMP7, ar fi implicate în apariția anoftalmiei. Potrivit lui Schilter , mai puțin de 40% dintre mutațiile genetice cunoscute ar explica anoftalmia. Pentru el, o mutație a genelor NF1, PAX6, SOX2 ar fi responsabilă de anoftalmie sau microftalmie. Ați descris o mutație a genei OTX2 asociată cu anoftalmie și microftalmie congenitală într-o familie chineză. Veronica a observat, la un pacient, o mutație a genei RAX2 responsabilă de anoftalmie unilaterală. Mutațiile genei TWIST în timpul delețiilor cromozomului 7 pot duce la disgenesie facială cu anoftalmie . Identificarea mutației unei alele a genei SIX6 la un pacient cu anoftalmie bilaterală a fost remarcată de Gallardo .
Fără cariotipare, nu suntem în măsură să atribuim aceste anoftalmii ereditare din studiul nostru unui anumit tip de mutație. Mai mult decât atât, sindromul Patau care este una dintre principalele etiologii nu poate fi reținut clinic la pacienții noștri.
Dintre toți acești factori etiologici, teoria mutației genetice pare să se potrivească cu cazurile noastre clinice, într-adevăr, aceste 3 cazuri provin din aceeași familie, dar și dintr-un cuplaj consangvin, deoarece ei înșiși provin din aceeași familie deoarece sunt verișori. Cu toate acestea, trebuie remarcat faptul că deficitul de vitamina A, expunerea la raze X, utilizarea abuzivă a solvenților, consumul de talidomină și infecțiile dobândite în timpul sarcinii ar fi, de asemenea, factori de mediu . Diagnosticul de anoftalmie poate fi pus pre sau postnatal pe baza unei combinații de semne clinice, imagistică și analiză genetică. Dufit , folosind noi tehnici imagistice (IRM fetală) a descris un caz de anoftalmie fetală asociată cu malformații multiple care a dus la întreruperea sarcinii.
La copii, tratamentul protetic prin conformații de mărime crescută, sau chiar tehnici de expansiune orbitală prin proteze gonflabile, este recomandat înainte de vârsta de 2 ani. Cu toate acestea, la adulți, acest tip de tratament devine inutil și este descurajat. Regimul terapeutic trebuie să includă o mărire a orbitei osoase prin diferite osteotomii asociate în același timp sau secundar cu refacerea sacului conjunctival prin grefă de mucoasă sau grefă dermo-epidermică. Apoi, restaurarea pleoapelor va avea loc prin reconstrucția, repoziționarea și corectarea malpoziției pe proteză . În țările noastre în curs de dezvoltare, acest tip de tratament greoi și uneori dezamăgitor este dificil de realizat de către echipe uneori nepregătite. Mai mult, nivelul de sărăcie al părinților limitează accesibilitatea acestora la tratament, cu consecințe estetice și o repercusiune socială majoră.
Concluzie
Anoftalmia este extrem de rară. Diagnosticul său implică investigarea atât a etiologiilor asociate, cât și a malformațiilor. Tratamentul său este greoi și neaccesibil uneori copiilor din clasa socială nefavorabilă, ceea ce duce la prejudecăți estetice. Formarea calificată pentru diagnosticul prenatal, gestionarea precoce de către specialiști calificați și o platformă tehnică adecvată sunt mijloacele esențiale pentru a reduce prejudiciul social.
Declarație etică
Acest raport de caz a urmat principiile Declarației de la Helsinki și a fost aprobat de Centrul Spitalului Universitar din Bouaké. Consimțământul a fost obținut de la părinții nou-născutului și nu există niciun conflict de interese cu privire la acest articol.
- Speeg-Schatz, Le Marec B. Erreurs génétiques et œil in: Flamment J, Storck D. Œil et pathologie générale Rapport de la soc Fr Ophtalmol. Masson ed, Paris; 1997. P27
- Guthoff R, Klein R, Lieb WE (2004) Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 242: 268-271.
- Mouriaux F, Audo I, Defoort-Dhellemmes S, Labalette P, Guilbert F, et al. (1997) . J Fr Ophtalmol 20: 583-591.
- Roth P, Roth A, Riethmuller D. Ophtalmologie fœtale: l’utilité de l’examen des yeux au cours de l’échographie anténatale. Ophtalmologie 1997; 11: 85-96.
- Romero Caballero MD, López Soler JA, Alcázar Cantos A (2002) . Arch Soc Esp Oftalmol 77: 571-574.
- Busby A, Dolk H, Collin R, Jones RB, Winter R (1998) Compilarea unui registru național de copii născuți cu anoftalmie/microftalmie în Anglia 1988-94. Arch Dis Child Fetal Neonatal Ed 79: F168-173.
- Araujo J E, Kawanami TE, Nardozza LM, Milani HJ. Oliveira PS, Monron AF (2012) Diagnosticul prenatal al anoftalmiei bilaterale prin ecografie 3D cu vedere „față inversă” și imagistică prin rezonanță magnetică. Taiwan J Obstet Gynecol 51: 616-619.
- Verma AS, Fitzpatrick DR (2007) Anoftalmie și microftalmie. Orphanet J Rare Dis 2: 47.
- Kouassi FX, Koffi KV, Safede K, Cochard C, Cochener B (2006) . J Fr Ophtalmol 29: e10.
- Diomande I.A, Toure.A, Koffi. K.V, Diomande.G F, Djiguimde.W P, N Habib, A. Ahnoux-zabsonre (2015) Anoftalmia și microftalmia gravă: un rezumat al problemelor asociate cu diagnosticul antenatal și rambursarea terapeutică în Subsahariană. Africa International Medical Case Reports Journal 8: 287-290.
- Stoll C, Dott B, Alembik Y, Roth MP (2012) Malformații asociate în rândul sugarilor cu anoftalmie și microftalmie. Birth Defects Res A Clin Mol Teratol 94: 147-152.12.
- Stricker M, Gola R (1990) L’anophtalmie congénitale Chirurgie plastique et réparatrice des paupières et de leurs annexes.Paris : Masson, 177-191.
- Morax.S (1998) Anophtalmies microphtalmies in: Adenis JP, Morax S. Morax S. Pathologie orbito-palpebrală. Rapport de la Soc Fr Ophtalmol. Paris: Masson. 635-645.
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, et al. (2003) Mutațiile în SOX2 cauzează anoftalmie. Nat Genet 33: 461-463.
- Lee B, Song H, Rizzoti K, Son Y, Yoon J, et al. (2013) Codul genomic pentru legarea Sox2 descoperă rolul său de reglementare în activarea Six3 în forebrain. Dev Biol 381: 491-501.
- Ye FX, Fan XQ (2012) . Zhonghua Yan Yan Ke Za Zhi 48: 1049-1052.
- Schilter KF, Reis LM, Schneider A, Bardakjian TM, Abdul-Rahman O, et al. (2013) Whole-genome copy number variation analysis in anophthalmia and microphthalmia. Clin Genet 84: 473-481.
- You T, Lv Y, Liu S, Li F, Li F, Zhao Y, et al. (2012) Noua mutație OTX2 asociată cu anoftalmie congenitală și microftalmie într-o familie chineză Han. Acta Ophthalmol 90: e501-502.
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, et al. (2004) Mutații în gena homeobox RAX umană la un pacient cu anoftalmie și sclerocornee. Hum Mol Genet 13: 315-322.
- Gallardo ME, Rodríguez De Córdoba S, Schneider AS, Dwyer MA, Ayuso C, et al. (2004) Analiza genei homeobox SIX6 de dezvoltare la pacienții cu anoftalmie/microftalmie. Am J Med Genet A 129A: 92-94.
- Dufit C, Baggio E, Ruban JM, Buenerd A, Hermier M (1999) Anoftalmie fetală. Problema diagnosticului prenatal: un raport de caz. J Fr Ophtalmol 22: 966-969.