Abstract
Introdução: A anoftemalia congênita é a ausência clínica do olho ao nascer. Ela resulta da falta de desenvolvimento ou regressão da vesícula óptica primária durante a vida embrionária. É rara e pode ser isolada ou associada a outros defeitos oculares ou gerais ao nascimento.
Observações: Relatamos os casos de três crianças da mesma família com idades compreendidas entre 4 meses e 8 anos com anoftalmias congênitas bilaterais. A sua história é marcada pela falta de consulta e de testes laboratoriais pré-natais. Exame oftalmológico objetivado, abertura das pálpebras com dois olhos, preenchimento da cavidade pelo tecido conjuntival em 3 casos sugestivos de anoftemia bilateral, confirmou a ultra-sonografia ocular.
Discussão: A anoftimia congênita é uma malformação rara. Ela pode ser isolada ou integrada a uma síndrome de malformação. O quadro clínico é em sua maioria unilateral. O diagnóstico é principalmente clínico e confirmado pela ultra-sonografia ocular. As causas são variadas, representadas por aberrações cromossômicas, mutações genéticas, intoxicações e infecções adquiridas durante a gravidez.
Conclusão: A descoberta de uma anoftemia congênita impõe uma revisão abrangente, a fim de buscar a etiologia. O apoio psicológico dos pais também continua sendo um foco importante.
Palavras-chave
Anofaltalmia, congênita, ultra-som ocular, aberrações cromossômicas
Introdução
Anofaltalmia é a falta clínica do olho. É uma anomalia oftalmológica extremamente rara. Existem numerosas etiologias, tais como aberrações cromossômicas, mutações genéticas, intoxicações e infecções. A anofalmia congênita pode ser isolada ou associada a síndromes polimorfológicas graves. É responsável, por um lado, por uma microorbit e uma hemiatrofia craniofacial homolateral e, por outro, por um fraco desenvolvimento dos apêndices que colocam problemas durante a adaptação protética. O diagnóstico tem sido melhorado pela imagem pré-natal e pelo manejo terapêutico, que envolve essencialmente próteses orbitais. Relatamos 3 casos de anoftemia congênita bilateral na mesma família.
Caso 1
É uma criança de 8 anos de idade, nascida a termo, do sexo masculino, recebida em consulta para uma diminuição bilateral da acuidade visual. Não foi possível a avaliação pré-natal biológica e orbito-cerebral por tomografia computadorizada. O exame oftalmológico clínico revelou ausência de percepção luminosa em ambos os olhos; anofaltalmia bilateral; os apêndices estão presentes e normais e na abertura das pálpebras, uma estrutura conjuntival preenchendo toda a cavidade orbital sem globo ocular. A tomografia órbito-cerebral solicitada confirmou a ausência do globo ocular com a presença de gordura residual, músculos oculomotores e atrofia do calibre do nervo óptico na cavidade orbital (Figura 1). O balanço geral foi normal. E as imagens de ressonância magnética não puderam ser realizadas considerando as dificuldades financeiras dos pais, assim como o equilíbrio genético (cariotipagem). O tratamento cirúrgico com colocação de uma prótese foi proposto mas não se materializou devido à falta de uma plataforma técnica adequada. (Figura 2)
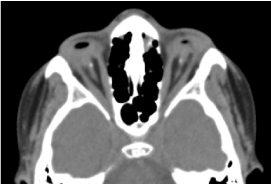
Figure.1. Tomografia computadorizada cerebral de órbita: ausência do globo ocular com gordura residual, músculos e atrofia do calibre do nervo otocerval na cavidade orbital.

Figure. 2. criança de 8 anos mostrando anofthalmos bilaterais na abertura das pálpebras
Caso 2
Criança masculina de 8 anos, na qual não foi realizada avaliação pré-natal (ecografia biológica e ultra-sonográfica). Foi recebido em consulta oftalmológica para uma diminuição bilateral da acuidade visual. O exame oftalmológico clínico encontrou ausência de percepção luminosa em ambos os olhos; uma anofalmia bilateral, apêndices sem particularidades e na abertura das pálpebras, uma estrutura conjuntival preenchendo a cavidade). O exame orbitocerebral solicitado confirmou a ausência do globo ocular com presença de gordura residual, músculos oculomotores e atrofia do calibre do nervo óptico na cavidade orbital (Figura 3). O exame geral desta criança foi normal. E a ressonância magnética, bem como o cariótipo, não puderam ser realizados pelas mesmas razões mencionadas anteriormente, bem como o manejo médico. (Figura 4)
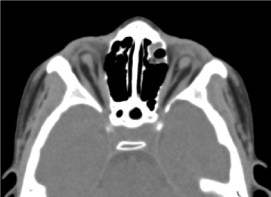
Figure.3Ausência do globo ocular com gordura residual, músculos e atrofia do calibre do nervo otocerval na cavidade orbital.
Caso 3
É o de uma menina de 4 meses de idade, nascida a termo e com bom desenvolvimento psicomotor e staturo-peso, recebida em consulta oftalmológica para diminuição bilateral da acuidade visual e oclusão permanente das pálpebras desde o nascimento. A história da criança é marcada por duas consultas pré-natais, mas sem exames biológicos e ultra-sonográficos pré-natais. O exame oftalmológico clínico encontrou ausência de rastreamento luminoso; anofaltalmia bilateral; aspectos normais dos apêndices e à abertura das pálpebras, um globo ocular em branco dando um aspecto microftalmológico, os detalhes da íris não são analisáveis e ausência de uma câmara anterior. A consulta pediátrica em busca de outras malformações retornou sem particularidade a nível clínico.
A tomografia órbito-cerebral solicitada confirmou a ausência de globo ocular com presença de gordura residual, músculos oculomotores e atrofia do calibre do nervo óptico na cavidade orbital (Figura 5). A ressonância magnética e o cariotipagem não puderam ser realizados; assim como o seu manejo médico (Figura 6).

Figure.4. Criança de 6 anos de idade com anoftimia bilateral na abertura das pálpebras.
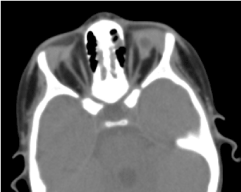
Figure. 5. Ausência de globo ocular com vesícula residual, músculos e atrofia do nervo otocerval na cavidade orbital.
 Figure.6. criança de 4 meses de idade; na abertura das pálpebras podemos notar, especialmente no olho direito um esboço de córnea, mas a íris não é individualizável.
Figure.6. criança de 4 meses de idade; na abertura das pálpebras podemos notar, especialmente no olho direito um esboço de córnea, mas a íris não é individualizável.
Discussão
Anoftalmia congênita ou ausência clínica do olho ao nascer é uma malformação rara. A sua incidência é variável. É estimada em menos de um caso a cada 10000 nascimentos para Mouriaux e Roth . Seria de cerca de 21,34 por 100.000 nascimentos, segundo um estudo espanhol e 10 por 100.000 nascimentos, segundo um estudo britânico. Para Aranjo , sua incidência é de 0,6 por 10000 nascimentos de crianças nascidas vivas. Esta diferença na incidência encontra a sua explicação na variabilidade da ocorrência desta malformação. A frequência da anoftemia não é influenciada por raça ou sexo. A anoftemalma pode ser isolada ou integrada em uma síndrome polimalformativa em um terço dos casos. Esta observação está em conformidade com as de vários autores. Assim, para Verna , tanto a anofalmia quanto a microftalmia podem ocorrer isoladamente ou ser integradas a uma síndrome de Patau (trissomia13) ou outras síndromes em cerca de um terço dos casos. Kouassi também relatou um caso de anoftalmias congênitas bilaterais durante a síndrome de Patau. Assim como Diomandé em Bouaké que relatou um caso de anoftemia congênita real associada a microftalmia grave em um recém-nascido sem qualquer outra anormalidade clínica observada.
Além disso, Stoll observou que 90% da anoftemia tinha malformações associadas. Portanto, na presença de qualquer anofalmia congênita ou microftalmia extrema, é importante realizar uma avaliação pediátrica completa em busca de anormalidades associadas, às vezes graves como anencefalia ou agenesia de todo o trato óptico. A anofalmia clínica é raramente bilateral. Investigações paraclínicas modernas como a tomografia computadorizada e a ressonância magnética tornam possível a investigação das malformações associadas. Em nossas observações, esses equilíbrios não puderam ser realizados dadas as dificuldades sócio-econômicas dos pais.
De acordo com Speeg – Schatz , existem três tipos de anoftalmitia congênita: anoftalmitia primária, anoftalmitia secundária e anoftalmitia secundária consecutiva como resultado da ausência de invaginação da fossa óptica, a completa cessação do desenvolvimento do tubo neural anterior ou a degeneração da vesícula óptica após a sua invaginação. Para Stricker , a anoftalmitia congênita poderia ser analisada e classificada de acordo com dois modos.
Do ponto de vista anatômico, seria necessário distinguir a verdadeira anoftalmitia de embriopatias caracterizadas pela ausência de qualquer contorno ocular. Somente o exame histológico confirmaria isto.
Do ponto de vista clínico, a anoftalmia clínica ou microftalmia extrema deve ser separada de outras microftalmia mais moderadas com prognóstico funcional às vezes melhor. Nos casos posteriores, há um globo ocular mais ou menos rudimentar (como nos nossos 3º casos clínicos) às vezes acompanhado de um cisto colobomatoso capaz de atuar como prótese expansiva fisiológica. Clinicamente, não há estrutura ocular, mas uma ultra-sonografia ocular, tomografia computadorizada e ressonância magnética mostrariam a presença de um esboço do globo ocular e do nervo óptico.
No entanto, Morax , notou que a verdadeira anofalmia não existia. Segundo ele, nos casos clínicos mais marcados, a neuroimagem mostraria a presença de um nervo óptico e de um pequeno esboço ocular. Em nossos dois primeiros casos clínicos, a ultra-sonografia ocular mostrou ausência de aspereza ocular. Entretanto, na ausência de resultados de tomografia e ressonância magnética, não podemos concluir formalmente que não há nervo óptico ocular nem nervo óptico. Pode ser hereditária por aberrações cromossômicas, em particular a trissomia do cromossomo 13 ou a trissomia do cromossomo 18. Neste caso, é a anoftemia com malformações cerebrais ou associada a uma displasia da linha mediana. Outras etiologias genéticas são representadas por mutações cromossômicas que podem ocorrer durante as deleções cromossômicas ou independentemente de qualquer aberração do cariótipo. As anomalias SOX2 são as principais. Fantes diagnosticou uma microdeleção associada a uma translocação t (3; 11) (q26-3 p11-2) e mutação do gene SOX2 em um paciente com anoftemia bilateral. Acredita-se que a haploinsuficiência de SOX2 seja responsável por anoftemia, microftalmia, displasia sépto-óptica e um defeito no desenvolvimento do cérebro anterior. Ye observou que o defeito SOX2 causaria um distúrbio múltiplo do sistema orgânico, incluindo a anoftalmia e a microftalmia. Outros genes, tais como PAX6, OTX2, CHV10, RAX2, SIX6, NF1, TWIST, SHH, BMP4 e BMP7, estariam envolvidos na ocorrência de anoftalmia. De acordo com Schilter, menos de 40% das mutações genéticas conhecidas explicariam a anoftalmia. Para ele, uma mutação dos genes NF1, PAX6, SOX2 seria responsável pela anoftalmia ou microftalmia. Você descreveu uma mutação do gene OTX2 associada à anoftalmia e microftalmia congênita em uma família chinesa. Veronica observou, em um paciente, uma mutação do gene RAX2 responsável pela anoftalmia unilateral. Mutações no gene TWIST durante as deleções do cromossomo 7 podem levar a disgenesia facial com anoftalmias. A identificação da mutação de um alelo do gene SIX6 em um paciente com anoftalmia bilateral foi notada por Gallardo .
Sem cariotipagem, não somos capazes de atribuir esses anofthalmos hereditários em nosso estudo a um tipo particular de mutação. Além disso, a síndrome de Patau que é uma das principais etiologias não pode ser retida clinicamente em nossos pacientes.
De todos esses fatores etiológicos, a teoria da mutação genética parece se encaixar em nossos casos clínicos, de fato, esses 3 casos vêm da mesma família, mas também de um acoplamento consanguíneo, pois eles mesmos vêm da mesma família, por serem primos. No entanto, deve-se notar que deficiência de vitamina A, exposição a raios X, uso abusivo de solvente, ingestão de talidomin e infecções adquiridas durante a gravidez também seriam fatores ambientais. O diagnóstico de anoftalmias pode ser feito pré ou pós-natal com base em uma combinação de sinais clínicos, imagens e análise genética. Dufit , utilizando novas técnicas de imagem (RM fetal) descreveu um caso de anoftemia fetal associada a múltiplas malformações levando à interrupção da gravidez.
Em crianças, o tratamento protético através do aumento de conformadores de tamanho, ou mesmo técnicas de expansão orbital por próteses infláveis, é recomendado antes dos 2 anos de idade. No entanto, em adultos, este tipo de tratamento torna-se desnecessário e desencorajado. O regime terapêutico deve incluir um alargamento da órbita óssea por diferentes osteotomias associadas ao mesmo tempo ou secundariamente à restauração do saco conjuntival por enxerto de mucosa ou enxerto dermo-epidérmico. Então, a restauração das pálpebras ocorrerá por reconstrução, reposicionamento e correção da malposição na prótese. Em nossos países em desenvolvimento, este tipo de tratamento pesado e às vezes decepcionante é difícil de ser realizado por equipes não treinadas. Além disso, o nível de pobreza dos pais limita a sua acessibilidade ao tratamento, com consequências estéticas e uma grande repercussão social.
Conclusão
Anofaltalmia é extremamente rara. Seu diagnóstico envolve a investigação tanto de etiologias associadas quanto de malformações. Seu tratamento é pesado e às vezes não acessível a crianças de classe social desfavorável, resultando em preconceito estético. A formação qualificada para o diagnóstico pré-natal, a gestão precoce por especialistas treinados e uma plataforma técnica adequada são os meios essenciais para reduzir os danos sociais.
Declaração ética
Estes casos relatados seguiram os princípios da Declaração de Helsinque e foram aprovados pelo Centro Hospitalar Universitário de Bouaké. O consentimento foi obtido dos pais do recém-nascido e não há conflito de interesses sobre este artigo.
- Speeg-Schatz, Le Marec B. Erreurs génétiques et œil in: Flamment J, Storck D. Œil et pathologie générale Rapport de la soc Fr. Ophtalmol. Masson ed, Paris; 1997. P27
- Guthoff R, Klein R, Lieb WE (2004) Congenital cystic eye. Graefes Arch Clin Exp Ophthalmol 242: 268-271.
- Mouriaux F, Audo I, Defoort-Dhellemmes S, Labalette P, Guilbert F, et al. (1997) . J Fr Ophtalmol 20: 583-591.
- Roth P, Roth A, Riethmuller D. Ophtalmologie fœtale: l’utilité de l’examen des yeux au cours de l’échographie anténatale. Ophtalmologie 1997; 11: 85-96.
- Romero Caballero MD, López Soler JA, Alcázar Cantos A (2002) . Arch Soc Esp Oftalmol 77: 571-574.
- Busby A, Dolk H, Collin R, Jones RB, Winter R (1998) Compilação de um registo nacional de bebés nascidos com anoftalmia/microftalmia na Inglaterra 1988-94. Arch Dis Child Fetal Neonatal Ed 79: F168-173.
- Araujo J E, Kawanami TE, Nardozza LM, Milani HJ. Oliveira PS, Monron AF (2012) Diagnóstico pré-natal de anoftemão bilateral por ultra-som 3D de “face reversa” e ressonância magnética. Taiwan J Obstet Gynecol 51: 616-619.
- Verma AS, Fitzpatrick DR (2007) Anoftalmias e microftalmia. Orphanet J Rare Dis 2: 47.
- Kouassi FX, Koffi KV, Safede K, Cochard C, Cochener B (2006) . J Fr Ophtalmol 29: e10.
- Diomande I.A, Toure.A, Koffi. K.V, Diomande.G F, Djiguimde.W P, N Habib, A. Ahnoux-zabsonre (2015) Anophthalmia and serious microphthalmia: a summary of the problems associated with antenatal diagnosis and therapeutic refunding in Sub-Saharan. Africa International Medical Case Reports Journal 8: 287-290.
- Stoll C, Dott B, Alembik Y, Roth MP (2012) Malformações associadas entre bebés com anoftalmias e microftalmia. Defeitos de Nascimento Res A Clin Mol Teratol 94: 147-152.12.
- Stricker M, Gola R (1990) L’anophtalmie congénitale Chirurgie plastique et réparatrice des paupières et de leurs annexes.Paris : Masson, 177-191.
- Morax.S (1998) Anophtalmies microphtalmies in: Adenis JP, Morax S. Pathologie orbito-palpébrale. Rapport de la Soc Fr. Ophtalmol. Paris: Masson. 635-645.
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, et al. (2003) As mutações em SOX2 causam anoftalmias. Nat Genet 33: 461-463.
- Lee B, Song H, Rizzoti K, Son Y, Yoon J, et al. (2013) Genomic code for Sox2 binding uncover its regulatory role in Six3 activation in the forebrain. Dev Biol 381: 491-501.
- Ye FX, Fan XQ (2012) . Zhonghua Yan Ke Za Zhi 48: 1049-1052.
- Schilter KF, Reis LM, Schneider A, Bardakjian TM, Abdul-Rahman O, et al. (2013) Whole-genome copy number variation analysis in anophthalmia and microphthalmia. Clin Genet 84: 473-481.
- You T, Lv Y, Liu S, Li F, Zhao Y, et al. (2012) Novel OTX2 mutation associated with congenital anophthalmia and microphthalmia in a Han Chinese family. Acta Ophthalmol 90: e501-502.
- Voronina VA, Kozhemyakina EA, O’Kernick CM, Kahn ND, Wenger SL, et al. (2004) Mutações no gene RAX homeobox humano em um paciente com anoftemia e esclerocornéia. Hum Mol Genet 13: 315-322.
- Gallardo ME, Rodríguez De Córdoba S, Schneider AS, Dwyer MA, Ayuso C, et al. (2004) Analysis of the developmental SIX6 homeobox gene in patients with anophthalmia/microphthalmia. Am J Med Genet A 129A: 92-94.
- Dufit C, Baggio E, Ruban JM, Buenerd A, Hermier M (1999) Fetal anophthalmos. Problema do diagnóstico pré-natal: um relato de caso. J Fr Ophtalmol 22: 966-969.