W rutynowej praktyce laboratoryjnej rozpoznanie cechy beta talasemii jest zwykle stawiane na podstawie charakterystycznych wyników oceny hemoglobiny oraz liczby i wskaźników krwinek czerwonych. W szczególności, odsetek hemoglobiny (Hb) A2 jest podwyższony, podczas gdy średnia objętość krwinki czerwonej (MCV) i/lub średnia hemoglobina (MCH) są obniżone. Liczba czerwonych krwinek jest zazwyczaj prawidłowa lub zwiększona, ale może być zmniejszona, jeśli u pacjenta występują inne przyczyny niedokrwistości.
Cechy beta talasemii (zwanej również beta talasemią minor lub stanem nosicielstwa beta talasemii) to łagodny, heterozygotyczny stan, który można odróżnić od cięższych zespołów beta talasemii (intermedia i major) na podstawie cech klinicznych i laboratoryjnych. Beta talasemia intermedia i major są związane z rosnącym nasileniem niedokrwistości, zależnością od transfuzji i splenomegalią, podczas gdy cechy te nie występują w przypadku cech beta talasemii. W ciężkiej postaci beta talasemii poziom hemoglobiny płodowej (Hb F) jest znacznie podwyższony z powodu braku Hb A, natomiast ilość Hb A2 może nie być zwiększona jak w przypadku cechy beta talasemii. Zatem laboratoryjne rozpoznanie cechy beta talasemii powinno być stosunkowo proste, prawda?
Nie zawsze. Poniższe dwa przypadki ilustrują typowe sytuacje, które mogą skomplikować diagnozę.
Przypadek 1: warunki konkurencyjne
Pacjentką była 28-letnia Afroamerykanka, u której stwierdzono zakażenie HIV-1 i która była poddawana wysoce aktywnej terapii antyretrowirusowej (HAART) z zastosowaniem schematu dolutegrawir/abakawir/lamiwudyna. Wyniki badań laboratoryjnych przedstawiono w tabeli 1.
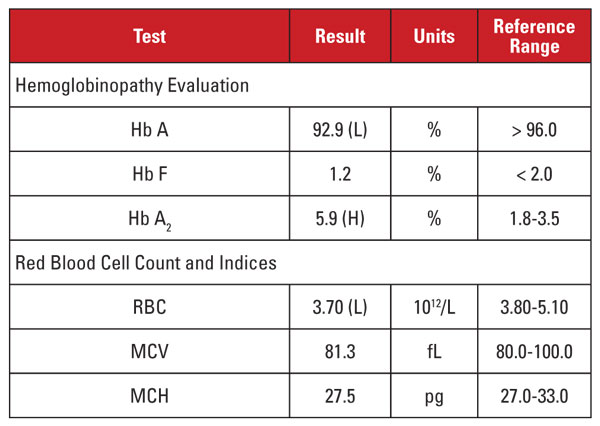
Ten przypadek przedstawiał ciekawą sytuację. Chociaż poziom Hb A2 był znacznie podwyższony, odpowiadający mu hemogram nie wykazywał mikrocytozy ani hipochromii. Kiedy wyniki badań laboratoryjnych nie dają podstaw do rozpoznania talasemii beta, warto rozważyć inne schorzenia, które wpływają na poziom Hb A2 i wskaźniki czerwonokrwinkowe. W tym przypadku ważną wskazówkę znaleziono w historii klinicznej pacjenta.
Stężenie Hb A2 ma tendencję do zwiększania się w stanach, które opóźniają dojrzewanie jądrowe prekursorów krwinek czerwonych. Stany te są również związane ze zwiększoną MCV.1,2 Najczęstszą przyczyną tego zjawiska jest niedokrwistość megaloblastyczna spowodowana niedoborem folianów i/lub witaminy B12. Podobne działanie ma jednak kilka leków hamujących syntezę kwasów nukleinowych, w tym klasa leków anty-HIV zwanych nukleozydowymi inhibitorami odwrotnej transkryptazy (NRTI). Lamiwudyna w schemacie HAART tego pacjenta jest lekiem z grupy NRTI.
Czy powinniśmy zatem stwierdzić, że wysoki poziom Hb A2 u tego pacjenta był spowodowany lekami anty-HIV, a pacjent nie miał cech talasemii beta? Nie tak szybko! Ogólnie rzecz biorąc, wzrost stężenia Hb A2 spowodowany stosowaniem NRTI (lub niedokrwistością megaloblastyczną) jest mniejszy niż ten obserwowany w przypadku cech talasemii beta.1,2 Ponadto wzrost ten jest proporcjonalny do ogólnego działania leku, co można w przybliżeniu określić na podstawie wzrostu MCV.1,2 U wielu pacjentów stosujących terapię NRTI MCV jest wyraźnie wysoka (często większa niż 120 fL), podczas gdy u naszego pacjenta MCV była w kierunku niskiego końca zakresu referencyjnego.
Dlatego nie byliśmy przekonani, że bardzo wysoki poziom Hb A2 u pacjenta był spowodowany wyłącznie terapią lamiwudyną. Podejrzewaliśmy, że pacjent miał konkurujące warunki, które zwiększały i zmniejszały MCV (tj. odpowiednio, cecha talasemii beta i terapia lamiwudyną), a oba przyczyniły się do wysokiego poziomu Hb A2. Poinformowaliśmy lekarza prowadzącego, że cecha talasemii beta jest prawdopodobna i może być potwierdzona przez analizę mutacji beta globiny. Późniejsze sekwencjonowanie genu ujawniło heterozygotyczną mutację beta globiny związaną z talasemią beta.
Przypadek 2: czynnik etniczny
Pacjentką była 35-letnia ciężarna Wietnamka, u której przeprowadzono badania przesiewowe w kierunku hemoglobinopatii. Wyniki badań laboratoryjnych przedstawiono w tabeli 2.
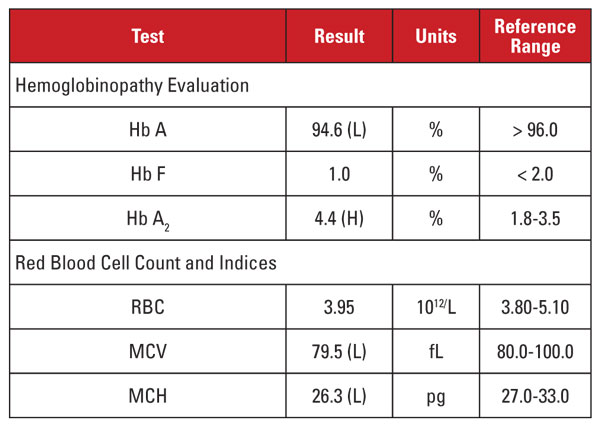
Ponownie, wysoki poziom Hb A2 był niezgodny z minimalną lub nieobecną mikrocytozą i hipochromią. W tym przypadku byliśmy w stanie wykluczyć wspólne czynniki zakłócające, takie jak niedobór folianów, witaminy B12 lub żelaza, zaburzenia czynności tarczycy lub efekty leków. W oparciu o pochodzenie etniczne pacjenta podejrzewaliśmy inną ważną przyczynę niezgodności: współdziedziczenie cechy talasemii beta z talasemią alfa minor. Kiedy pacjent dziedziczy oba te schorzenia, MCV i MCH często normalizują się, ponieważ łańcuchy globiny alfa i beta są obecne w stosunkowo zrównoważonych ilościach w rozwijających się krwinkach czerwonych.2
Zaleciliśmy wykonanie analiz genetycznych w celu zbadania mutacji talasemii alfa i beta, aby w pełni ocenić ryzyko dla ciężarnej matki i płodu. Późniejsze sekwencjonowanie globiny beta ujawniło heterozygotyczną mutację beta-plus talasemii, podczas gdy analiza delecji globiny alfa ujawniła heterozygotyczną delecję dwóch genów w regionie Azji Południowo-Wschodniej (SEA), zgodną z -/αα alfa talasemią minor. Na podstawie tych wyników wskazana była analiza genetyczna ojca w celu oceny ryzyka odziedziczenia przez płód klinicznie ciężkiego zespołu talasemii.
Talasemie alfa i beta
Talasemie należą do najczęstszych zaburzeń dziedziczonych u ludzi. Są one bardzo rozpowszechnione tam, gdzie malaria była endemiczna, a obecnie są powszechne w każdej części świata z powodu migracji populacji ludzkich. Talasemie są powodowane przez mutacje, które zmniejszają ekspresję genu globiny w prekursorach czerwonych krwinek. Zaburzenia te są klasyfikowane według genów globiny, które są zmutowane (na przykład, alfa versus beta) oraz według ciężkości choroby, która jest związana z tym, czy mutacje są dziedziczone w sposób heterozygotyczny czy homozygotyczny/złożony heterozygotyczny. Główną postacią hemoglobiny u dorosłych jest Hb A, tetramer złożony z dwóch łańcuchów globiny alfa i dwóch łańcuchów globiny beta. W talasemii alfa ekspresja globiny alfa jest niewystarczająca i występuje odpowiedni nadmiar łańcuchów globiny beta.
Ten wzór jest odwrócony w talasemii beta. W warunkach niedoboru i braku równowagi łańcuchów globiny, zawartość hemoglobiny w krwinkach czerwonych jest zmniejszona, co skutkuje mikrocytozą i hipochromią. Ponadto, nadmiar łańcuchów globiny alfa lub beta tworzy niestabilne tetramery, które powodują hemolizę. Talasemię alfa i beta rozróżnia się na podstawie ilości mniejszej hemoglobiny dorosłego człowieka Hb A2, tetrameru dwóch łańcuchów globiny alfa i dwóch łańcuchów globiny delta. Hb A2 jest zwiększona w talasemii beta, ponieważ względny brak globiny beta pozwala na włączenie do hemoglobiny większej ilości łańcuchów delta.
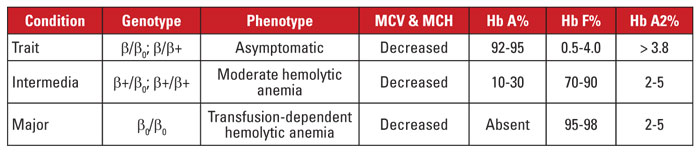
Talasemia beta jest spowodowana mutacjami w locus genu globiny beta na chromosomie 11.3,4 Większość mutacji to małe substytucje nukleotydów, insercje lub delecje, chociaż duże delecje są identyfikowane w rzadkich przypadkach. W zależności od mutacji, ekspresja globiny beta jest zmniejszona częściowo (talasemia beta-plus) lub całkowicie (talasemia beta-zero). Cecha beta talasemii jest spowodowana mutacją heterozygotyczną. Stan ten jest bezobjawowy i charakteryzuje się podwyższonym stężeniem Hb A2, mikrocytozą krwinek czerwonych i brakiem istotnych niedokrwistości hemolitycznych. Z kolei talasemia beta major (niedokrwistość Cooleya) jest spowodowana homozygotyczną mutacją beta-zero. Ocena hemoglobiny wykazuje przewagę Hb F, brak Hb A oraz prawidłową lub zwiększoną ilość Hb A2. Talasemia beta major charakteryzuje się ciężką, zależną od transfuzji niedokrwistością hemolityczną, z wynikającą z tego splenomegalią i deformacjami kostnymi. Nawracające transfuzje prowadzą do przeładowania żelazem, co jest główną przyczyną zachorowalności i śmiertelności. Beta talasemia intermedia jest klinicznie niejednorodna, z większością objawów związanych z umiarkowaną niedokrwistością hemolityczną. Zależność od transfuzji jest rzadka, ponieważ ekspresja globiny beta nie jest nieobecna. Ze względu na dużą liczbę mutacji związanych z talasemią beta, diagnostyka genetyczna wymaga zazwyczaj sekwencjonowania genów. Zespoły talasemii beta podsumowano w tabeli 3.
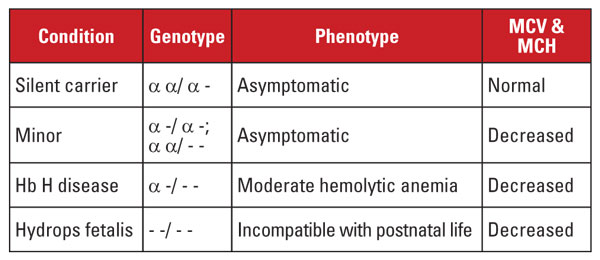
Talasemia alfa jest spowodowana mutacjami w locus genu globiny alfa na chromosomie 16.5 Geny globiny alfa są zduplikowane na chromosomie 16, więc normalny osobnik ma cztery kopie. Najczęstszymi mutacjami są duże delecje, które wpływają na jeden lub oba geny w locus. Opisano dość ograniczoną liczbę delecji, z przewagą w różnych populacjach; na przykład, duże mutacje, które usuwają oba geny alfa są bardzo powszechne u ludzi z południowo-wschodniej Azji. Mutacje te są również powszechne w populacjach śródziemnomorskich, ale są rzadkie u osób o afrykańskim pochodzeniu etnicznym. W przeciwieństwie do tego, delecje pojedynczych genów globiny alfa są szeroko rozpowszechnione. Utrata jednego z czterech genów globiny alfa jest określana jako stan cichego nosicielstwa.
Stan ten jest bezobjawowy i zwykle związany z prawidłowymi wskaźnikami czerwonokrwinkowymi. Utrata dwóch genów globiny alfa jest określana mianem talasemii alfa minor. Stan ten może wystąpić z powodu homozygotycznej delecji jednego genu lub heterozygotycznej delecji dwóch genów. Liczba czerwonych krwinek i wskaźniki są nie do odróżnienia od cech talasemii beta, ale poziom Hb A2 jest prawidłowy. Utrata trzech genów alfa jest określana jako choroba Hb H. (Hb H jest tetramerem łańcuchów globiny beta.) Choroba Hb H jest zwykle umiarkowaną niedokrwistością hemolityczną związaną ze splenomegalią i zmianami kostnymi. Zależność od transfuzji jest rzadka.
Utrata wszystkich czterech genów alfa jest określana jako Hb Barts hydrops fetalis (Hb Barts jest tetramerem łańcuchów gamma globiny płodowej). Stan ten jest nie do pogodzenia z życiem postnatalnym. Hb H i Hb Barts mogą być zidentyfikowane w ocenie hemoglobiny i odgrywają rolę w postnatalnej diagnostyce talasemii alfa.5
Diagnostyka genetyczna jest zwykle przeprowadzana za pomocą testów łańcuchowej reakcji polimerazy z lukami (PCR), które są ukierunkowane na wspólne delecje. Sekwencjonowanie genu globiny alfa jest również dostępne w celu wykrycia rzadkich mutacji. Zespoły talasemii alfa są podsumowane w Tabeli 4.
Kiedy potrzebne są badania genetyczne
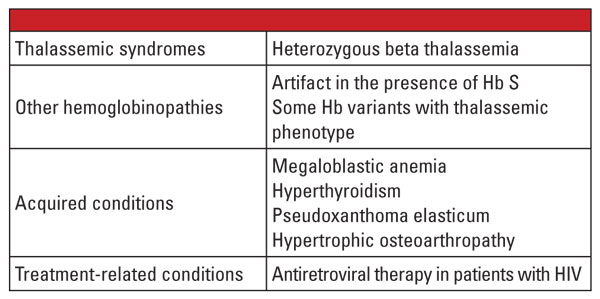
W podsumowaniu, heterozygotyczne formy talasemii alfa i beta występują bardzo często i są często identyfikowane w laboratorium klinicznym. Ocena hemoglobiny i inne rutynowe dane kliniczne i laboratoryjne są zazwyczaj wystarczające do postawienia diagnozy. Badania genetyczne nie są konieczne w większości przypadków, ale mogą mieć decydujące znaczenie w kontekście planowania rodziny. Jeśli mutacje talasemii zostaną zidentyfikowane u obojga rodziców, może istnieć znaczące ryzyko, że płód odziedziczy ciężką talasemię. W przypadku wodogłowia płodowego prawdopodobne jest obumarcie płodu, a matka jest narażona na znaczne ryzyko powikłań ciąży.5 Dlatego ważne jest, aby być świadomym zmiennych, które komplikują laboratoryjną diagnostykę talasemii i rozpoznać, kiedy wskazane jest wykonanie potwierdzających badań genetycznych. Na przykład, jak przedstawiono w pierwszym opisie przypadku, rozpoznanie cech talasemii beta może stanowić wyzwanie, jeśli współistnieją schorzenia wpływające na poziom Hb A2. Czynniki, o których wiadomo, że zwiększają stężenie Hb A2, wymieniono w tabeli 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biology, clinical relevance and a possible target for ameliorating sickle cell disease. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Gene test review. Alpha-thalassemia. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH recommendations for the measurement of haemoglobin A2. Int J Lab Hematol. 2012;34(1):1-13.