In de routine-laboratoriumpraktijk wordt de diagnose van bèta-thalassemie meestal gesteld op grond van karakteristieke bevindingen bij de hemoglobine-evaluatie en het aantal en de indices van de rode bloedcellen. In het bijzonder is het percentage hemoglobine (Hb) A2 verhoogd, terwijl het gemiddelde corpusculaire volume (MCV) en/of het gemiddelde corpusculaire hemoglobine (MCH) zijn verlaagd. Het aantal rode bloedcellen is meestal normaal of verhoogd, maar kan verlaagd zijn als de patiënt andere oorzaken van bloedarmoede heeft.
Bèta-thalassemie trait (ook wel bèta-thalassemie minor of bèta-thalassemie carrier state genoemd) is een goedaardige, heterozygote aandoening die kan worden onderscheiden van de ernstiger bèta-thalassemie syndromen (intermedia en major) door klinische en laboratoriumkenmerken. Bèta-thalassemie intermedia en major worden geassocieerd met toenemende ernst van de anemie, afhankelijkheid van transfusie en splenomegalie, terwijl deze kenmerken afwezig zijn bij bèta-thalassemie trait. Bij ernstige bèta-thalassemie is het niveau van foetale hemoglobine (Hb F) duidelijk verhoogd door de afwezigheid van Hb A, en is de hoeveelheid Hb A2 mogelijk niet verhoogd zoals bij bèta-thalassemie-treatit. De laboratoriumdiagnose van beta thalassemie zou dus relatief eenvoudig moeten zijn, juist?
Niet altijd. De volgende twee gevallen illustreren veel voorkomende situaties die de diagnose kunnen compliceren.
Geval 1: concurrerende omstandigheden
De patiënte was een 28-jarige Afro-Amerikaanse vrouw die positief was voor HIV-1-infectie en die zeer actieve antiretrovirale therapie (HAART) onderging met een dolutegravir/abacavir/lamivudineregime. De laboratoriumresultaten worden weergegeven in tabel 1.
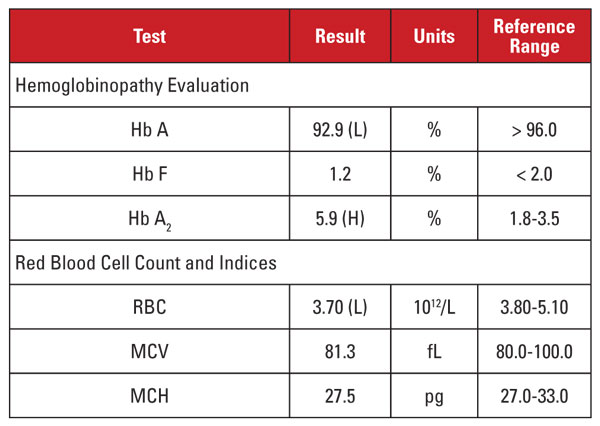
Dit geval vertoonde een interessante situatie. Hoewel het Hb A2-gehalte significant verhoogd was, toonde het bijbehorende hemogram geen microcytose of hypochromie aan. Wanneer de laboratoriumkenmerken niet wijzen op de diagnose bèta-thalassemie, is het zinvol andere aandoeningen te overwegen die van invloed zijn op de Hb A2-spiegel en de rode bloedcelindices. In dit geval werd een belangrijke aanwijzing gevonden in de klinische voorgeschiedenis van de patiënt.
Hb A2 niveaus hebben de neiging te stijgen in omstandigheden die de nucleaire rijping van rode cel precursors vertragen. Deze condities worden ook geassocieerd met een verhoogd MCV.1,2 De meest voorkomende oorzaak van dit fenomeen is megaloblastaire anemie als gevolg van foliumzuur- en/of vitamine B12-deficiëntie. Verschillende medicijnen die de nucleïnezuursynthese remmen hebben echter een soortgelijk effect, waaronder de klasse van anti-HIV medicijnen die nucleoside reverse transcriptase inhibitors (NRTI’s) worden genoemd. Lamivudine in het HAART-schema van deze patiënt is een NRTI-medicijn.
Moeten we dan concluderen dat het hoge Hb A2-gehalte van deze patiënt te wijten was aan de anti-HIV-medicatie, en dat de patiënt geen bèta-thalassemie-trek had? Niet zo snel! In het algemeen is de toename van Hb A2 als gevolg van NRTI (of megaloblastaire anemie) minder groot dan die welke wordt gezien bij bèta-thalassemie.1,2 Bovendien zijn de toenames meestal evenredig met de algemene effecten van het geneesmiddel, die kunnen worden benaderd door toenames in MCV.1,2 Bij veel patiënten die NRTI-therapie krijgen, is het MCV duidelijk hoog (vaak meer dan 120 fL), terwijl bij onze patiënt het MCV aan de lage kant van het referentiebereik lag.
Daarom waren wij er niet van overtuigd dat het zeer hoge Hb A2-niveau van de patiënt uitsluitend te wijten was aan lamivudinetherapie. Wij vermoedden dat de patiënt een concurrerende aandoening had die het MCV verhoogde en verlaagde (d.w.z. respectievelijk de bèta thalassemie eigenschap en de lamivudinetherapie), waarbij beide bijdroegen aan een hoge Hb A2-spiegel. We meldden de aanvragende arts dat beta-thalassemie waarschijnlijk was, en kon worden bevestigd door analyse van betaglobinemutatie. Daaropvolgende gensequencing onthulde een heterozygote beta globine mutatie geassocieerd met beta-zero thalassemie.
Geval 2: etniciteitsfactor
De patiënte was een 35-jarige zwangere Vietnamese vrouw die werd gescreend op hemoglobinopathie. De laboratoriumresultaten zijn weergegeven in tabel 2.
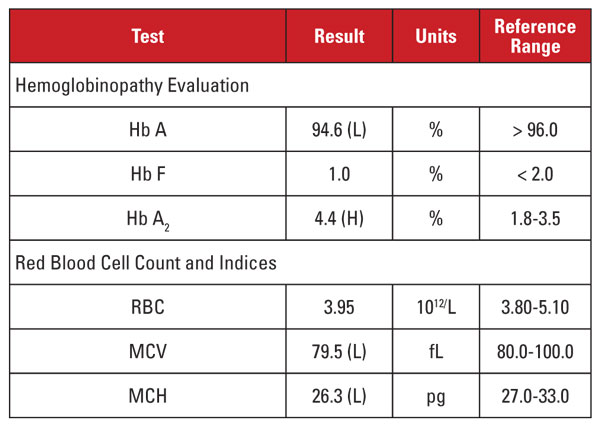
Ook hier was het hoge Hb A2-niveau niet in overeenstemming met minimale of afwezige microcytose en hypochromie. In dit geval konden we veel voorkomende verstorende factoren uitsluiten, zoals foliumzuur-, vitamine B12- of ijzertekort, schildklierdisfunctie of medicijneffecten. Op basis van de etniciteit van de patiënt, vermoedden we een andere belangrijke oorzaak van discordantie: beta thalassemia trait co-inherited met alpha thalassemia minor. Wanneer een patiënt beide aandoeningen erft, normaliseren het MCV en het MCH vaak omdat alfa- en betaglobineketens in relatief evenwichtige hoeveelheden aanwezig zijn in zich ontwikkelende rode bloedcellen.2
Wij adviseerden genetische analyses om te testen op alfa- en beta- thalassemiemutaties, om het risico voor de zwangere moeder en de foetus volledig te kunnen beoordelen. Latere sequencing van beta-globine toonde een heterozygote beta-plus thalassemie mutatie, terwijl analyse voor alfa-globine deleties een heterozygote Zuidoost-Aziatische (SEA) twee-gen deletie aan het licht bracht, consistent met -/αα alpha thalassemia minor. Op basis van deze bevindingen was genetische analyse van de vader aangewezen om het risico voor de foetus op overerving van een klinisch ernstig thalassemie syndroom te beoordelen.
Alpha en beta thalassemieën
Thalassemieën behoren tot de meest voorkomende erfelijke aandoeningen bij de mensheid. Zij komen veel voor op plaatsen waar malaria endemisch is geweest, en zijn nu algemeen in elk deel van de wereld als gevolg van de migratie van de menselijke bevolking. Thalassemieën worden veroorzaakt door mutaties die de expressie van het globinegen in de voorlopers van de rode bloedcellen verminderen. De aandoeningen worden ingedeeld naar de gemuteerde globinegenen (bijvoorbeeld alfa versus bèta) en naar de ernst van de ziekte, die afhangt van het feit of de mutaties heterozygoot of homozygoot/compound heterozygoot worden overgeërfd. De belangrijkste volwassen vorm van hemoglobine is Hb A, een tetramer van twee alfa- en twee betaglobineketens. Bij alfa-thalassemie is de expressie van alfa-globine deficiënt en is er een overeenkomstige overmaat aan bèta-globineketens.
Dit patroon is omgekeerd bij bèta-thalassemie. In de setting van globine keten deficiëntie en onbalans, is de rode cel hemoglobinegehalte verlaagd, wat resulteert in microcytose en hypochromie. Bovendien vormen overtollige alfa- of bèta-globineketens onstabiele tetrameren die hemolyse veroorzaken. Alfa- en bèta-thalassemie worden onderscheiden door de hoeveelheid van het minder belangrijke volwassen hemoglobine Hb A2, een tetrameer van twee alfa- en twee delta-globineketens. Hb A2 is verhoogd bij bèta-thalassemie omdat door het relatieve gebrek aan bèta-globine meer delta-ketens in hemoglobine kunnen worden opgenomen.
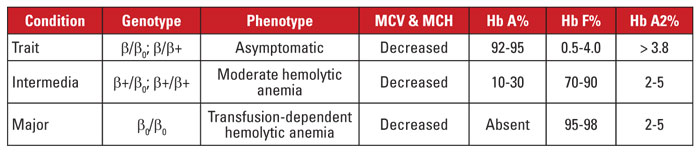
Bèta-thalassemie wordt veroorzaakt door mutaties in de locus van het bèta-globinegen op chromosoom 11.3,4 De meeste mutaties zijn kleine nucleotide-substituties, inserties of deleties, hoewel in zeldzame gevallen grote deleties worden vastgesteld. Afhankelijk van de mutatie is de expressie van bèta-globine gedeeltelijk (bèta-plus thalassemie) of volledig (bèta-nul thalassemie) gereduceerd. Bèta-thalassemie wordt veroorzaakt door een heterozygote mutatie. Deze aandoening is asymptomatisch en wordt gekenmerkt door een verhoogd Hb A2, microcytose van de rode bloedcellen, en geen significante hemolytische anemie. Bèta-thalassemie major (Cooley’s anemie) daarentegen wordt veroorzaakt door homozygote bèta-nul mutaties. Hemoglobine-evaluatie toont een predominantie van Hb F, afwezig Hb A, en normaal of verhoogd Hb A2. Bèta-thalassemie major wordt gekenmerkt door ernstige, transfusie-afhankelijke hemolytische anemie, met als gevolg splenomegalie en misvormingen van de botten. Recurrente transfusie leidt tot ijzeroverbelasting, die een belangrijke oorzaak is van morbiditeit en mortaliteit. Bèta-thalassemie intermedia is klinisch heterogeen, waarbij de meeste symptomen verband houden met matige hemolytische anemie. Transfusie-afhankelijkheid is ongewoon omdat bèta-globine-expressie niet afwezig is. Door het grote aantal mutaties dat geassocieerd is met bèta-thalassemie, is voor genetische diagnose meestal gensequencing nodig. De bèta-thalassemie-syndromen zijn samengevat in tabel 3.
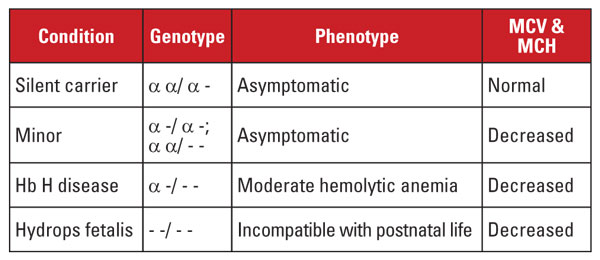
Alfalassemie wordt veroorzaakt door mutaties in de locus van het alfaglobinegen op chromosoom 16.5 De alfaglobinegenen worden op chromosoom 16 gedupliceerd, zodat een normaal individu vier kopieën heeft. De meest voorkomende mutaties zijn grote deleties die één of beide genen op de locus beïnvloeden. Er is een vrij beperkt aantal deleties beschreven, met prevalentie in verschillende populaties; grote mutaties waarbij beide alfa-genen worden verwijderd, komen bijvoorbeeld zeer vaak voor bij mensen van Zuidoost-Aziatische etniciteit. Deze mutaties komen ook veel voor bij mediterrane bevolkingsgroepen, maar zijn zeldzaam bij mensen van Afrikaanse etniciteit. Daarentegen zijn deleties van afzonderlijke alfaglobinegenen wijdverbreid. Het verlies van één van de vier alfaglobinegenen wordt een “silent carrier state” genoemd.
Deze aandoening is asymptomatisch en gaat gewoonlijk gepaard met normale rode bloedcel indices. Het verlies van twee alfa-globinegenen wordt alfa-thalassemie minor genoemd. Deze aandoening kan optreden als gevolg van homozygote één-gen deleties of heterozygote twee-gen deleties. Het aantal rode cellen en de indices zijn niet te onderscheiden van bèta-thalassemie trait, maar het Hb A2-gehalte is normaal. Het verlies van drie alfa-genen wordt de Hb H-ziekte genoemd. (Hb H is een tetramer van beta globine ketens.) Hb H ziekte is typisch een matige hemolytische anemie geassocieerd met splenomegalie en botveranderingen. Transfusie-afhankelijkheid is ongewoon.
Het verlies van alle vier alfa genen wordt aangeduid als Hb Barts hydrops fetalis (Hb Barts is een tetramer van foetale gamma globine ketens). Deze aandoening is onverenigbaar met het postnatale leven. Hb H en Hb Barts kunnen bij hemoglobinetests worden geïdentificeerd en spelen een rol bij de postnatale diagnose van alfa-thalassemie.5
Genetische diagnose wordt gewoonlijk verricht door gap-polymerasekettingreactietests (PCR) die gericht zijn op de veel voorkomende deleties. sequencing van het alfaglobinegen is ook beschikbaar om zeldzame mutaties op te sporen. De alfa-thalassemie-syndromen zijn samengevat in tabel 4.
Wanneer genetische tests nodig zijn
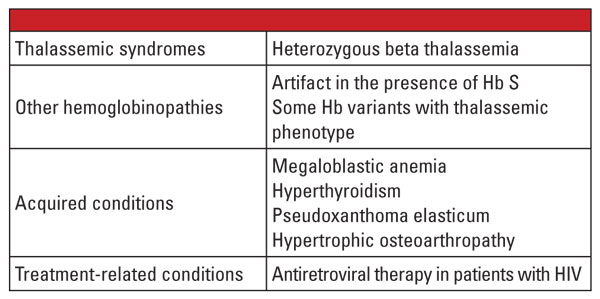
Concluderend kan worden gesteld dat de heterozygote vormen van alfa- en bèta-thalassemie zeer vaak voorkomen en frequent in het klinische laboratorium worden geïdentificeerd. Hemoglobine-evaluatie en andere routinematige klinische en laboratoriumgegevens zijn gewoonlijk voldoende voor de diagnose. Genetische tests zijn in de meeste gevallen niet nodig, maar kunnen van cruciaal belang zijn in het kader van de gezinsplanning. Als bij beide ouders thalassemiemutaties worden vastgesteld, kan er een aanzienlijk risico bestaan dat de foetus ernstige thalassemie erft. In het geval van hydrops fetalis is foetale sterfte waarschijnlijk en loopt de moeder een aanzienlijk risico op zwangerschapscomplicaties.5 Daarom is het belangrijk zich bewust te zijn van variabelen die de laboratoriumdiagnose van thalassemieën bemoeilijken, en te onderkennen wanneer bevestigend genetisch onderzoek geïndiceerd is. Bijvoorbeeld, zoals aangetoond in de eerste casusrapportage, kan de diagnose van beta thalassemie trait een uitdaging zijn als er co-existente aandoeningen zijn die het niveau van Hb A2 beïnvloeden. Factoren waarvan bekend is dat ze het Hb A2-gehalte kunnen verhogen, staan vermeld in tabel 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biologie, klinische relevantie en een mogelijk doelwit voor de verbetering van sikkelcelziekte. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Gene test review. Alfa-thalassemie. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH recommendations for the measurement of haemoglobin A2. Int J Lab Hematol. 2012;34(1):1-13.