Nella pratica di laboratorio di routine, la diagnosi di tratto di beta talassemia è di solito fatta dai risultati caratteristici nella valutazione dell’emoglobina e la conta e gli indici dei globuli rossi. In particolare, la percentuale di emoglobina (Hb) A2 è elevata, mentre il volume corpuscolare medio dei globuli rossi (MCV) e/o l’emoglobina corpuscolare media (MCH) sono diminuiti. La conta dei globuli rossi è di solito normale o aumentata, ma può essere diminuita se il paziente ha altre cause di anemia.
Il tratto di beta talassemia (chiamato anche beta talassemia minore o stato di portatore di beta talassemia) è una condizione eterozigote benigna che può essere distinta dalle sindromi beta talassemia più gravi (intermedia e maggiore) da caratteristiche cliniche e di laboratorio. Beta talassemia intermedia e maggiore sono associati con crescente gravità di anemia, dipendenza da trasfusioni e splenomegalia, mentre queste caratteristiche sono assenti nel tratto di beta talassemia. Nella beta talassemia grave, il livello di emoglobina fetale (Hb F) è notevolmente aumentato a causa dell’assenza di Hb A, e la quantità di Hb A2 può non essere aumentata come nel tratto di beta talassemia. Quindi la diagnosi di laboratorio del tratto di beta talassemia dovrebbe essere relativamente semplice, giusto?
Non sempre. I seguenti due casi illustrano situazioni comuni che possono complicare la diagnosi.
Caso 1: condizioni concorrenti
Il paziente era una donna afroamericana di 28 anni che era positiva all’infezione da HIV-1 e stava seguendo una terapia antiretrovirale altamente attiva (HAART) con un regime dolutegravir/abacavir/lamivudina. I risultati di laboratorio sono mostrati nella Tabella 1.
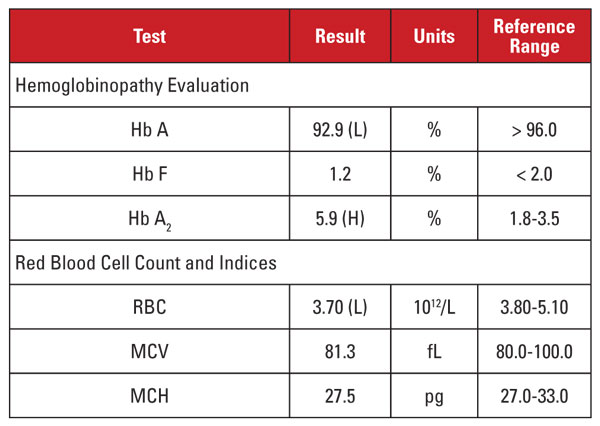
Questo caso presentava una situazione interessante. Anche se il livello di Hb A2 era significativamente elevato, l’emogramma corrispondente non dimostrava microcitosi o ipocromia. Quando le caratteristiche di laboratorio sono discordanti per una diagnosi di tratto di beta talassemia, è utile considerare altre condizioni che influenzano il livello di Hb A2 e gli indici dei globuli rossi. In questo caso, un indizio importante è stato trovato nella storia clinica del paziente.
I livelli di Hb A2 tendono ad aumentare nelle condizioni che ritardano la maturazione nucleare dei precursori dei globuli rossi. Queste condizioni sono anche associate ad un aumento del MCV.1,2 La causa più comune di questo fenomeno è l’anemia megaloblastica dovuta alla carenza di folati e/o vitamina B12. Tuttavia, diversi farmaci che inibiscono la sintesi dell’acido nucleico hanno un effetto simile, compresa la classe di farmaci anti-HIV chiamati inibitori della trascrittasi inversa nucleosidica (NRTI). La lamivudina nel regime HAART di questo paziente è un farmaco NRTI.
Dovremmo quindi concludere che l’alto livello di Hb A2 di questo paziente era dovuto al farmaco anti-HIV e che il paziente non aveva un tratto di beta-talassemia? Non così in fretta! In generale, gli aumenti di Hb A2 dovuti agli NRTI (o all’anemia megaloblastica) sono inferiori a quelli osservati nel tratto di beta talassemia.1,2 Inoltre, gli aumenti tendono ad essere proporzionali agli effetti generali del farmaco, che possono essere approssimati dagli aumenti di MCV.1,2 In molti pazienti in terapia con NRTI, l’MCV è marcatamente alto (spesso superiore a 120 fL), mentre nel nostro paziente, l’MCV era verso l’estremità bassa del range di riferimento.
Quindi, non eravamo convinti che il livello molto alto di Hb A2 del paziente fosse dovuto esclusivamente alla terapia con lamivudina. Sospettavamo che il paziente avesse condizioni concorrenti che aumentavano e diminuivano l’MCV (cioè il tratto di beta-talassemia e la terapia con lamivudina, rispettivamente), e che entrambi contribuissero a un alto livello di Hb A2. Abbiamo riferito al medico ordinante che il tratto di beta-talassemia era probabile, e potrebbe essere confermato dall’analisi della mutazione della beta globina. Il successivo sequenziamento del gene ha rivelato una mutazione eterozigote della beta globina associata alla beta talassemia.
Caso 2: fattore etnico
La paziente era una donna vietnamita di 35 anni incinta che veniva sottoposta a screening per l’emoglobinopatia. I risultati di laboratorio sono mostrati nella tabella 2.
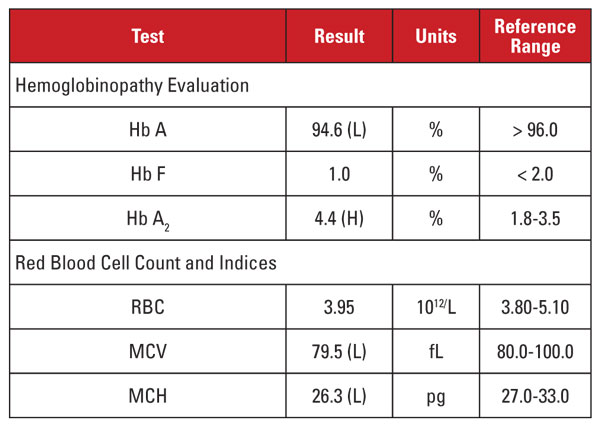
Ancora una volta, l’alto livello di Hb A2 era discordante con microcitosi e ipocromia minime o assenti. In questo caso, siamo stati in grado di escludere i comuni fattori di confondimento come la carenza di folati, vitamina B12 o ferro, la disfunzione tiroidea o gli effetti dei farmaci. Sulla base dell’etnia del paziente, abbiamo sospettato un’altra importante causa di discordanza: il tratto di beta-talassemia ereditato insieme alla alfa-talassemia minore. Quando un paziente eredita entrambe queste condizioni, l’MCV e l’MCH spesso si normalizzano perché le catene globiniche alfa e beta sono presenti in quantità relativamente bilanciate nei globuli rossi in via di sviluppo.2
Abbiamo raccomandato analisi genetiche per testare le mutazioni alfa e beta talassemia, al fine di valutare pienamente il rischio per la madre incinta e il feto. Il successivo sequenziamento della beta globina ha rivelato una mutazione eterozigote beta-plus talassemia, mentre l’analisi per le delezioni dell’alfa globina ha rivelato una delezione eterozigote Southeast Asian (SEA) a due geni, coerente con -/αα alfa talassemia minore. Sulla base di questi risultati, l’analisi genetica del padre era indicata per valutare il rischio per il feto di ereditare una sindrome di talassemia clinicamente grave.
Talassemie alfa e beta
Talassemie sono tra i disturbi ereditati più comuni nel genere umano. Sono altamente prevalenti dove la malaria è stata endemica, e sono ora comuni in ogni parte del mondo a causa della migrazione delle popolazioni umane. Le talassemie sono causate da mutazioni che riducono l’espressione del gene della globina nei precursori dei globuli rossi. I disturbi sono classificati dai geni della globina che sono mutati (per esempio, alfa contro beta) e dalla gravità della malattia, che è legata al fatto che le mutazioni sono ereditate in modo eterozigote o omozigote/compound eterozigote. La principale forma adulta di emoglobina è l’Hb A, un tetramero di due catene alfa e due beta globiniche. Nella talassemia alfa, l’espressione della globina alfa è carente e c’è un eccesso corrispondente di catene beta globiniche.
Questo modello è invertito nella talassemia beta. Nell’impostazione della carenza e dello squilibrio delle catene globiniche, il contenuto di emoglobina dei globuli rossi è diminuito, con conseguente microcitosi e ipocromia. Inoltre, le catene alfa o beta globiniche in eccesso formano tetrameri instabili che causano emolisi. Alfa e beta talassemia si distinguono per la quantità di emoglobina minore degli adulti Hb A2, un tetramero di due catene alfa e due delta globine. Hb A2 è aumentato in beta talassemia perché la relativa mancanza di beta globina permette più catene delta da incorporare in emoglobina.
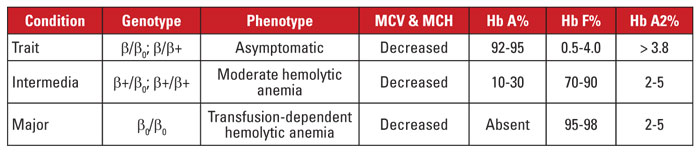
Beta talassemia è causata da mutazioni nel locus del gene beta globina sul cromosoma 11.3,4 La maggior parte delle mutazioni sono piccole sostituzioni nucleotidiche, inserzioni o delezioni, anche se grandi delezioni sono identificati in rari casi. A seconda della mutazione, l’espressione della beta globina è ridotta parzialmente (talassemia beta-plus) o completamente (talassemia beta-zero). Il tratto di beta talassemia è causato da una mutazione eterozigote. Questa condizione è asintomatica ed è caratterizzata da un aumento di Hb A2, microcitosi dei globuli rossi e nessuna anemia emolitica significativa. Al contrario, la beta talassemia major (anemia di Cooley) è causata da mutazioni omozigoti beta-zero. La valutazione dell’emoglobina rivela una predominanza di Hb F, Hb A assente e Hb A2 normale o aumentata. La beta talassemia major è caratterizzata da un’anemia emolitica grave e dipendente dalle trasfusioni, con conseguente splenomegalia e deformità ossee. La trasfusione ricorrente porta al sovraccarico di ferro, che è una causa principale di morbilità e mortalità. La beta talassemia intermedia è clinicamente eterogenea, con la maggior parte dei sintomi legati all’anemia emolitica moderata. La dipendenza da trasfusioni non è comune perché l’espressione della beta globina non è assente. A causa del gran numero di mutazioni associate alla beta talassemia, la diagnosi genetica richiede tipicamente il sequenziamento del gene. Le sindromi di beta talassemia sono riassunte nella tabella 3.
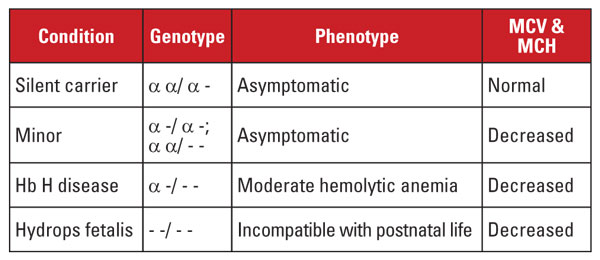
L’alfa talassemia è causata da mutazioni nel locus del gene alfa globina sul cromosoma 16.5 I geni alfa globina sono duplicati sul cromosoma 16, quindi un individuo normale ha quattro copie. Le mutazioni più comuni sono grandi delezioni che colpiscono uno o entrambi i geni del locus. È stato descritto un numero abbastanza limitato di delezioni, con prevalenza in diverse popolazioni; per esempio, grandi mutazioni che cancellano entrambi i geni alfa sono molto comuni nelle persone di etnia sud-est asiatica. Queste mutazioni sono anche prevalenti nelle popolazioni mediterranee, ma sono rare nelle persone di etnia africana. Al contrario, le delezioni di singoli geni alfa globinici hanno una distribuzione diffusa. La perdita di uno dei quattro geni alfa globinici è indicata come uno stato di portatore silenzioso.
Questa condizione è asintomatica e di solito è associata a indici dei globuli rossi normali. La perdita di due geni alfa globina è definita alfa talassemia minore. Questa condizione può verificarsi a causa di delezioni omozigoti di un gene o eterozigoti di due geni. La conta dei globuli rossi e gli indici sono indistinguibili dal tratto di beta talassemia, ma il livello di Hb A2 è normale. La perdita di tre geni alfa è chiamata malattia Hb H. (Hb H è un tetramero di catene beta globiniche.) La malattia Hb H è tipicamente un’anemia emolitica moderata associata a splenomegalia e cambiamenti ossei. La dipendenza da trasfusioni non è comune.
La perdita di tutti e quattro i geni alfa è definita Hb Barts hydrops fetalis (Hb Barts è un tetramero di catene gamma globiniche fetali). Questa condizione è incompatibile con la vita postnatale. Hb H e Hb Barts possono essere identificati nelle valutazioni dell’emoglobina e giocano un ruolo nella diagnosi postnatale dell’alfa talassemia.5
La diagnosi genetica è solitamente compiuta dai saggi della reazione a catena della polimerasi (PCR) che mirano alle delezioni comuni. Il sequenziamento del gene dell’alfa globina è disponibile anche per rilevare mutazioni rare. Le sindromi di alfa talassemia sono riassunte nella tabella 4.
Quando è necessario un test genetico
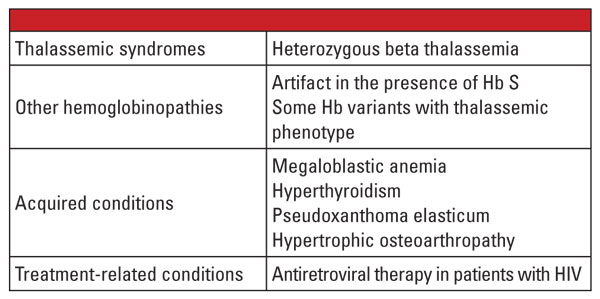
In conclusione, le forme eterozigoti di alfa e beta talassemia sono molto comuni e vengono identificate frequentemente nel laboratorio clinico. La valutazione dell’emoglobina e altri dati clinici e di laboratorio di routine sono solitamente sufficienti per la diagnosi. Il test genetico non è necessario nella maggior parte dei casi, ma può essere criticamente importante nel contesto della pianificazione familiare. Se le mutazioni della talassemia sono identificate in entrambi i genitori, ci può essere un rischio significativo per il feto di ereditare la talassemia grave. Nel caso di idrope fetale, la morte del feto è probabile e c’è un rischio significativo per la madre per le complicazioni della gravidanza.5 Pertanto, è importante essere consapevoli delle variabili che complicano la diagnosi di laboratorio di talassemie, e riconoscere quando è indicato un test genetico di conferma. Per esempio, come mostrato nel primo caso, la diagnosi di tratto di beta talassemia può essere difficile se ci sono condizioni coesistenti che influenzano il livello di Hb A2. I fattori che sono noti per aumentare l’Hb A2 sono elencati nella tabella 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biologia, rilevanza clinica e un possibile obiettivo per migliorare la malattia falciforme. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-talassemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-talassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Gene test review. Alfa-talassemia. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH recommendations for the measurement of haemoglobin A2. Int J Lab Hematol. 2012;34(1):1-13.