A rutin laboratóriumi gyakorlatban a béta-thalassaemia trait diagnózisát általában a hemoglobinértékelés és a vörösvértestszám és -indexek jellegzetes leletei alapján állítják fel. Különösen a hemoglobin (Hb) A2 százalékos aránya emelkedett, míg a vörösvértestek átlagos korpuszkuláris térfogata (MCV) és/vagy az átlagos korpuszkuláris hemoglobin (MCH) csökkent. A vörösvérsejtszám általában normális vagy emelkedett, de csökkenhet, ha a betegnek a vérszegénység más okai is vannak.
A béta-talasszémia trait (más néven béta-talasszémia minor vagy béta-talasszémia hordozó állapot) jóindulatú, heterozigóta állapot, amely klinikai és laboratóriumi jellemzők alapján megkülönböztethető a súlyosabb béta-talasszémia szindrómáktól (intermedia és major). A béta-talasszémia intermedia és major a vérszegénység növekvő súlyosságával, transzfúziófüggőséggel és splenomegáliával jár, míg a béta-talasszémia trait esetében ezek a jellemzők hiányoznak. Súlyos béta-talasszémiában a magzati hemoglobin (Hb F) szintje a Hb A hiánya miatt jelentősen megnövekszik, és a Hb A2 mennyisége nem feltétlenül emelkedik, mint béta-talasszémia-traitban. Tehát a béta-talasszémia trait laboratóriumi diagnózisának viszonylag egyszerűnek kell lennie, igaz?
Nem mindig. A következő két eset olyan gyakori helyzeteket illusztrál, amelyek megnehezíthetik a diagnózist.
1. eset: versengő körülmények
A beteg egy 28 éves afroamerikai nő volt, aki HIV-1-fertőzésre pozitív volt, és dolutegravir/abakavir/lamivudin kezelésben részesült magas aktivitású antiretrovirális terápiában (HAART). A laboratóriumi eredményeket az 1. táblázat tartalmazza.
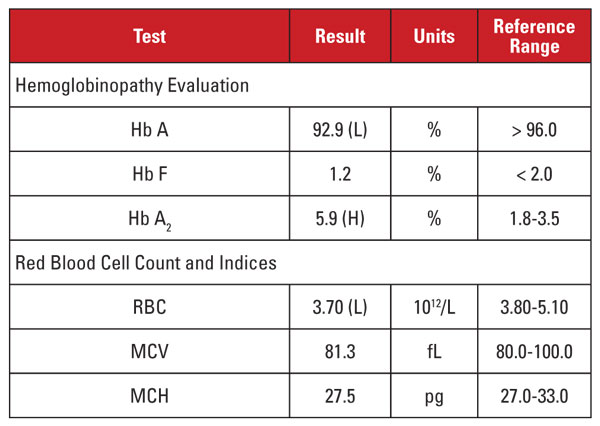
Ez az eset érdekes helyzetet mutatott. Bár a Hb A2-szint szignifikánsan emelkedett volt, a megfelelő hemogram nem mutatott mikrocitózist vagy hypochromiát. Ha a laboratóriumi jellemzők nem egyeznek meg a béta thalassemia trait diagnózisával, hasznos megfontolni a Hb A2-szintet és a vörösvértest-indexeket befolyásoló egyéb állapotokat. Ebben az esetben fontos támpontot találtunk a beteg klinikai anamnézisében.
A Hb A2-szintek hajlamosak emelkedni olyan körülmények között, amelyek késleltetik a vörösvértest-prekurzorok nukleáris érését. Ezek az állapotok megnövekedett MCV-vel is járnak.1,2 E jelenség leggyakoribb oka a folsav- és/vagy B12-vitaminhiány miatt kialakuló megaloblasztos anémia. Azonban számos, a nukleinsavszintézist gátló gyógyszer hasonló hatással bír, beleértve a HIV-ellenes gyógyszerek nukleozid reverz transzkriptáz gátlóknak (NRTI-k) nevezett osztályát. A beteg HAART-kezelésében szereplő lamivudin NRTI-gyógyszer.
Azt a következtetést kell tehát levonnunk, hogy ennek a betegnek a magas Hb A2-szintje a HIV-ellenes gyógyszereknek köszönhető, és a betegnek nem volt béta-talasszémiás vonása? Ne olyan gyorsan! Általában az NRTI (vagy megaloblasztos vérszegénység) okozta Hb A2 emelkedés kisebb mértékű, mint a béta-thalassaemia trait esetén tapasztaltak.1,2 Ráadásul az emelkedés általában arányos a gyógyszer általános hatásával, amit az MCV növekedésével lehet megközelíteni.1,2 Sok NRTI-terápiában részesülő betegnél az MCV kimondottan magas (gyakran 120 fL-nél nagyobb), míg betegünknél az MCV a referenciatartomány alsó vége felé volt.
Ezért nem voltunk meggyőződve arról, hogy a beteg nagyon magas Hb A2-szintje kizárólag a lamivudin-terápiának köszönhető. Azt gyanítottuk, hogy a betegnek egymással versengő állapotai voltak, amelyek növelték és csökkentették az MCV-t (azaz a béta-talasszémiás jelleg és a lamivudin-terápia), és mindkettő hozzájárult a magas Hb A2-szinthez. Jeleztük a megrendelő orvosnak, hogy a béta-talasszémiás jelleg valószínűsíthető, és ezt béta-globinmutáció-elemzéssel meg lehet erősíteni. A későbbi génszekvenálás heterozigóta béta-globinmutációt mutatott ki, amely a béta-zéró thalassemiához társult.
2. eset: etnikai tényező
A beteg egy 35 éves terhes vietnami nő volt, akit hemoglobinopathiára vizsgáltak. A laboratóriumi leleteket a 2. táblázat tartalmazza.
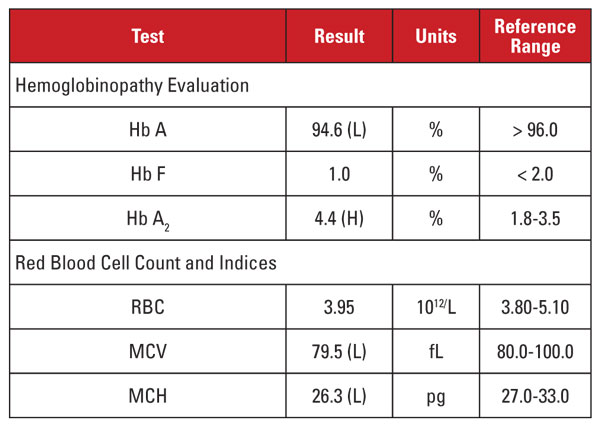
A magas Hb A2-szint ismét diszkordáns volt a minimális vagy hiányzó mikrocitózissal és hipokrómiával. Ebben az esetben ki tudtuk zárni az olyan gyakori zavaró tényezőket, mint a folsav-, B12-vitamin- vagy vashiány, pajzsmirigy-alulműködés vagy gyógyszerhatás. A beteg etnikai hovatartozása alapján a diszkordancia másik fontos okára gyanakodtunk: az alfa thalassemia minorral együtt öröklődő béta thalassemia traitra. Ha a beteg mindkét állapotot örökli, az MCV és az MCH gyakran normalizálódik, mivel az alfa- és béta globinláncok viszonylag kiegyensúlyozott mennyiségben vannak jelen a fejlődő vörösvértestekben.2
A terhes anya és a magzat kockázatának teljes körű felmérése érdekében genetikai vizsgálatokat javasoltunk az alfa- és béta-talasszémia mutációinak vizsgálatára. Az ezt követő béta globin szekvenálás heterozigóta béta-plusz thalassaemia mutációt mutatott ki, míg az alfa globin deléciók elemzése heterozigóta délkelet-ázsiai (SEA) két gén deléciót mutatott ki, ami összhangban van a -/αα alfa thalassaemia minorral. Ezen eredmények alapján az apa genetikai elemzése javallt annak felmérésére, hogy a magzatra nézve fennáll-e a klinikailag súlyos thalassemia-szindróma öröklésének kockázata.
Alfa- és béta-thalassémiák
A thalassémiák az emberiség leggyakoribb öröklődő rendellenességei közé tartoznak. Nagyon elterjedtek ott, ahol a malária endémiás volt, és az emberi populációk vándorlása miatt ma már a világ minden részén gyakoriak. A talasszémiákat olyan mutációk okozzák, amelyek csökkentik a globin gén kifejeződését a vörösvértestek előfutáraiban. A rendellenességeket a mutálódott globin gének (például alfa versus béta) és a betegség súlyossága szerint osztályozzák, ami attól függ, hogy a mutációk heterozigóta vagy homozigóta/összetett heterozigóta módon öröklődnek-e. A betegség súlyossága a mutációkat a génváltozatoktól függ. A hemoglobin fő felnőttkori formája a Hb A, amely két alfa és két béta globinláncból álló tetramer. Az alfa-talasszémiában az alfa-globin expressziója hiányos, és ennek megfelelően a béta-globin láncok túlsúlyban vannak.
A béta-talasszémiában ez a minta megfordul. Globinlánchiány és egyensúlyhiány esetén a vörösvérsejtek hemoglobintartalma csökken, ami mikrocitózist és hipokrómia kialakulását eredményezi. Ezenkívül a felesleges alfa- vagy béta-globinláncok instabil tetramereket alkotnak, amelyek hemolízist okoznak. Az alfa- és béta-talasszémiát a kisebb felnőttkori hemoglobin, a Hb A2 mennyisége alapján különböztetjük meg, amely két alfa- és két delta globinláncból álló tetramer. A béta-talasszémiában a Hb A2 mennyisége megnövekszik, mivel a béta globin relatív hiánya miatt több delta lánc épül be a hemoglobinba.
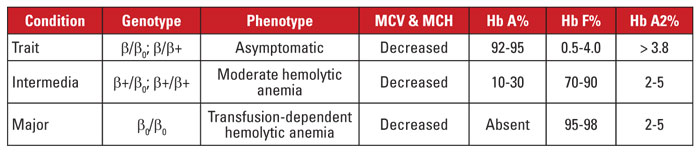
A béta-talasszémiát a 11. kromoszómán található béta globin gén lókuszában található mutációk okozzák.3,4 A legtöbb mutáció kis nukleotid szubsztitúció, inszerció vagy deléció, bár ritka esetekben nagy deléciók is előfordulnak. A mutációtól függően a béta globin expressziója részben (béta-plusz thalassemia) vagy teljesen (béta-nulla thalassemia) csökken. A béta-talasszémiás vonulatot heterozigóta mutáció okozza. Ez az állapot tünetmentes, és fokozott Hb A2, vörösvérsejt-mikrocitózis és nem jelentős hemolitikus anémia jellemzi. Ezzel szemben a béta-talasszémia major (Cooley-anémia) homozigóta béta-nulla mutáció okozta. A hemoglobinvizsgálat a Hb F túlsúlyát, a Hb A hiányát és normális vagy emelkedett Hb A2-t mutat. A béta thalassemia majorra súlyos, transzfúziófüggő hemolitikus anémia jellemző, amely splenomegáliával és csontdeformitásokkal jár. Az ismétlődő transzfúzió vastúlterheléshez vezet, ami a morbiditás és a mortalitás egyik fő oka. A béta thalassemia intermedia klinikailag heterogén, a legtöbb tünet a mérsékelt hemolitikus vérszegénységhez kapcsolódik. A transzfúziófüggőség nem ritka, mivel a béta globin expressziója nem hiányzik. A béta-talasszémiához társuló mutációk nagy száma miatt a genetikai diagnózis általában génszekvenálást igényel. A béta-talasszémia szindrómáit a 3. táblázat foglalja össze.
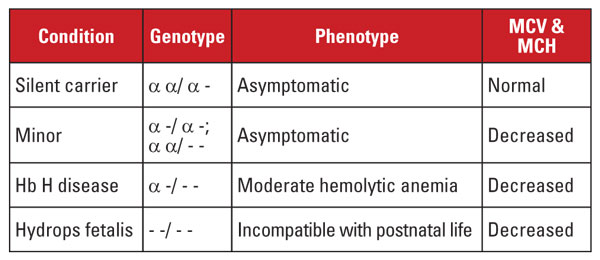
Az alfa-talasszémiát a 16-os kromoszómán található alfa-globin génlokusz mutációi okozzák.5 Az alfa-globin gének a 16-os kromoszómán duplikálódnak, így egy normális egyénnek négy példánya van. A leggyakoribb mutációk nagyméretű deléciók, amelyek a génlókusz egyik vagy mindkét génjét érintik. Meglehetősen korlátozott számú deléciót írtak le, amelyek előfordulási gyakorisága a különböző populációkban eltérő; például a délkelet-ázsiai etnikumú embereknél nagyon gyakoriak azok a nagyméretű mutációk, amelyek mindkét alfa-gént törlik. Ezek a mutációk a mediterrán népességben is gyakoriak, de az afrikai etnikumúaknál ritkák. Ezzel szemben az egyes alfa globin gének deléciói széles körben elterjedtek. A négy alfa-globin gén egyikének elvesztését csendes hordozói állapotnak nevezik.
Ez az állapot tünetmentes, és általában normális vörösvértest-indexekkel jár. Két alfa globin gén elvesztését alfa thalassemia minornak nevezzük. Ez az állapot előfordulhat homozigóta egy gén deléciója vagy heterozigóta két gén deléciója miatt. A vörösvérsejtszám és az indexek nem különböztethetők meg a béta-talasszémia jellegétől, de a Hb A2 szint normális. A három alfa gén elvesztését Hb H betegségnek nevezzük. (A Hb H a béta globinláncok tetramerje.) A Hb H betegség jellemzően mérsékelt hemolitikus vérszegénység, amelyhez splenomegália és csontelváltozások társulnak. A transzfúziófüggőség nem ritka.
A mind a négy alfa gén elvesztését Hb Barts hydrops fetalisnak nevezik (a Hb Barts a magzati gamma globin láncok tetramerje). Ez az állapot a születés utáni élettel összeegyeztethetetlen. A Hb H és a Hb Barts azonosítható a hemoglobinvizsgálatok során, és szerepet játszik az alfa-thalasszémia posztnatális diagnózisában.5
A genetikai diagnózis általában a közös deléciókat célzó rés-polimeráz láncreakció (PCR) vizsgálatokkal történik. A ritka mutációk kimutatására az alfa globin gén szekvenálása is rendelkezésre áll. Az alfa-thalasszémiás szindrómákat a 4. táblázat foglalja össze.
Mikor van szükség genetikai vizsgálatra
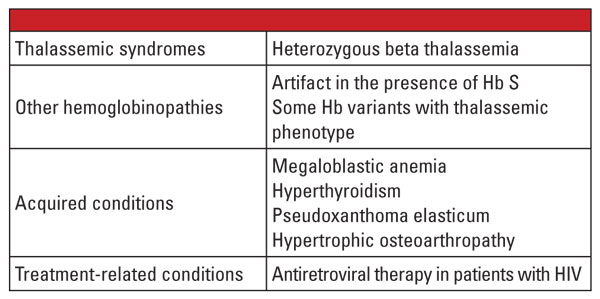
Végeredményben az alfa- és béta-thalasszémia heterozigóta formái nagyon gyakoriak, és gyakran azonosítják őket a klinikai laboratóriumban. A hemoglobin értékelése és más rutin klinikai és laboratóriumi adatok általában elegendőek a diagnózis felállításához. A genetikai vizsgálat a legtöbb esetben nem szükséges, de a családtervezés szempontjából kritikusan fontos lehet. Ha mindkét szülőnél talasszémiás mutációkat azonosítanak, a magzatra nézve jelentős kockázatot jelenthet a súlyos talasszémia öröklése. Hydrops fetalis esetén a magzat elhalása valószínű, és az anyára nézve a terhesség szövődményeinek jelentős kockázata áll fenn.5 Ezért fontos, hogy tisztában legyünk a thalassémiák laboratóriumi diagnózisát bonyolító változókkal, és felismerjük, mikor van szükség megerősítő genetikai vizsgálatra. Például, amint azt az első esetről szóló beszámoló is mutatja, a béta-talasszémiás jelleg diagnózisa kihívást jelenthet, ha olyan társbetegségek állnak fenn, amelyek befolyásolják a Hb A2 szintjét. Az 5.6. táblázatban
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biológia, klinikai jelentőség és a sarlósejtes betegség javításának lehetséges célpontja. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Géntesztek áttekintése. Alfathalasszémia. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH ajánlások a hemoglobin A2 mérésére. Int J Lab Hematol. 2012;34(1):1-13.