Rutiininomaisessa laboratoriokäytännössä beeta-talassemiataipumuksen diagnoosi tehdään yleensä hemoglobiiniarvion ja punasolujen määrän ja indeksien tyypillisten löydösten perusteella. Erityisesti hemoglobiini (Hb) A2:n prosenttiosuus on koholla, kun taas punasolujen keskimääräinen korpuskulaarinen tilavuus (MCV) ja/tai keskimääräinen korpuskulaarinen hemoglobiini (MCH) ovat pienentyneet. Punasolujen määrä on yleensä normaali tai suurentunut, mutta se voi olla pienentynyt, jos potilaalla on muita anemian syitä.
Beta-talassemian piirre (jota kutsutaan myös nimellä beeta-talassemia minor tai beeta-talassemian kantajatila) on hyvänlaatuinen, heterotsygoottinen tila, joka voidaan kliinisten ja laboratorio-ominaisuuksien perusteella erottaa vakavammista beeta-talassemian oireyhtymistä (intermedia ja major). Beetatalassemia intermedia ja major liittyvät anemian vaikeutumiseen, verensiirtoriippuvuuteen ja splenomegaliaan, kun taas beetatalassemian ominaisuudessa nämä piirteet puuttuvat. Vaikeassa beetatalassemiassa sikiön hemoglobiinin (Hb F) määrä on selvästi lisääntynyt Hb A:n puuttumisen vuoksi, eikä Hb A2:n määrä välttämättä ole lisääntynyt kuten beetatalassemian ominaisuudessa. Joten beeta-talassemiataipumuksen laboratoriodiagnoosin pitäisi olla suhteellisen suoraviivainen, eikö niin?
Ei aina. Seuraavat kaksi tapausta havainnollistavat yleisiä tilanteita, jotka voivat vaikeuttaa diagnoosia.
Tapaus 1: kilpailevat olosuhteet
Potilas oli 28-vuotias afroamerikkalainen nainen, jolla oli HIV-1-infektio ja joka sai erittäin aktiivista antiretroviraalista terapiaa (HAART), jossa käytettiin dolutegraviiri/abakaviiri/lamivudiini-hoitoa. Laboratoriolöydökset on esitetty taulukossa 1.
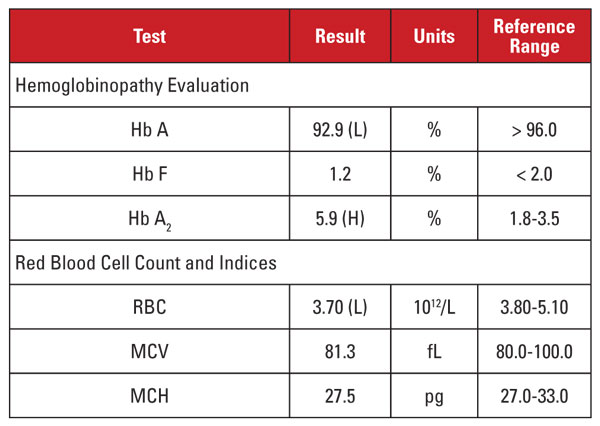
Tässä tapauksessa oli mielenkiintoinen tilanne. Vaikka Hb A2 -arvo oli merkittävästi koholla, vastaava hemogrammi ei osoittanut mikrosytoosia tai hypokromiaa. Kun laboratorio-ominaisuudet ovat ristiriidassa beeta-talassemiatriitin diagnoosin kanssa, on hyödyllistä harkita muita tiloja, jotka vaikuttavat Hb A2 -tasoon ja punasoluindekseihin. Tässä tapauksessa tärkeä vihje löytyi potilaan kliinisestä anamneesista.
Hb A2 -tasoilla on taipumus nousta olosuhteissa, jotka viivästyttävät punasolujen esiasteiden ydinkypsymistä. Näihin tiloihin liittyy myös lisääntynyt MCV.1,2 Tämän ilmiön yleisin syy on folaatin ja/tai B12-vitamiinin puutteesta johtuva megaloblastinen anemia. Useilla nukleiinihapposynteesiä estävillä lääkkeillä on kuitenkin samanlainen vaikutus, mukaan lukien HIV-lääkkeiden ryhmä, jota kutsutaan nukleosidikäänteistranskriptaasin estäjiksi (NRTI-lääkkeet). Tämän potilaan HAART-hoidossa oleva lamivudiini on NRTI-lääkitys.
Onko siis pääteltävä, että tämän potilaan korkea Hb A2 -taso johtui HIV-lääkityksestä, eikä potilaalla ollut beeta-talassemia-piirrettä? Ei niin nopeasti! Yleensä NRTI:n (tai megaloblastisen anemian) aiheuttama Hb A2:n nousu on vähäisempää kuin beeta-talassemiataipumuksen yhteydessä.1,2 Lisäksi nousu on yleensä verrannollinen lääkkeen kokonaisvaikutuksiin, joita voidaan approksimoida MCV:n nousulla.1,2 Monilla NRTI-hoitoa saavilla potilailla MCV on selvästi korkea (usein yli 120 fL), kun taas potilaallamme MCV oli kohti viitealueen alarajaa.
Siten emme olleet vakuuttuneita siitä, että potilaan erittäin korkea Hb A2 -taso johtui yksinomaan lamivudiinihoidosta. Epäilimme, että potilaalla oli kilpailevia tiloja, jotka suurensivat ja pienensivät MCV:tä (eli beetatalassemiataipumus ja lamivudiinihoito), ja molemmat vaikuttivat korkeaan Hb A2 -tasoon. Ilmoitimme tilaavalle lääkärille, että beetatalassemiataipumus oli todennäköinen, ja se voitaisiin vahvistaa beetaglobiinimutaatioanalyysillä. Myöhemmin suoritettu geenisekvensointi paljasti heterotsygoottisen beetaglobiinimutaation, joka liittyi beeta-taalassemiaan.
Tapaus 2: etnisyystekijä
Potilas oli 35-vuotias raskaana oleva vietnamilaisnainen, jota oltiin seulomassa hemoglobinopatian vuoksi. Laboratoriolöydökset on esitetty taulukossa 2.
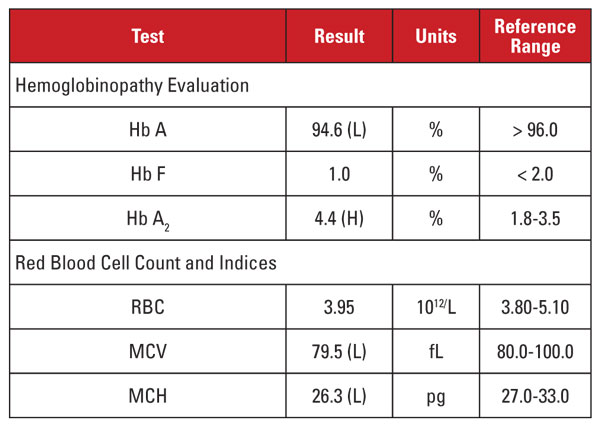
Korkea Hb A2 -taso ei sopinut yhteen minimaalisen tai puuttuvan mikrosytoosin ja hypokromian kanssa. Tässä tapauksessa pystyimme sulkemaan pois yleiset sekoittavat tekijät, kuten folaatin, B12-vitamiinin tai raudan puutteen, kilpirauhasen toimintahäiriön tai lääkkeiden vaikutukset. Potilaan etnisen alkuperän perusteella epäilimme toista merkittävää epäsuhtaisuuden syytä: beeta-talassemia-ominaisuutta, joka periytyy yhdessä alfa-talassemia minorin kanssa. Kun potilas perii molemmat näistä tiloista, MCV- ja MCH-arvot normalisoituvat usein, koska alfa- ja beetaglobiiniketjuja on kehittyvissä punasoluissa suhteellisen tasapainoisesti.2
Suosittelimme geneettisiä analyysejä alfa- ja beeta-talassemian mutaatioiden testaamiseksi, jotta voitaisiin täysin arvioida riski raskaana olevalle äidille ja sikiölle. Myöhemmin suoritettu beetaglobiinin sekvensointi paljasti heterotsygoottisen beeta-plus-talassemiamutaation, kun taas alfaglobiinin deleetioiden analyysi paljasti heterotsygoottisen kaakkois-aasialaisen (SEA) kahden geenin deleetion, joka sopi yhteen -/αα alfa-talassemia minorin kanssa. Näiden löydösten perusteella isän geneettinen analyysi oli aiheellinen sen arvioimiseksi, onko sikiöllä riski periä kliinisesti vaikea talassemiaoireyhtymä.
Alfa- ja beeta-talassemiat
Talassemiat ovat ihmiskunnan yleisimpiä perinnöllisiä sairauksia. Ne ovat erittäin yleisiä siellä, missä malaria on ollut endeeminen, ja ne ovat nykyään yleisiä kaikkialla maailmassa ihmisväestön muuttoliikkeen vuoksi. Talassemiat johtuvat mutaatioista, jotka vähentävät globiinigeenin ilmentymistä punasolujen esiasteissa. Sairaudet luokitellaan sen mukaan, mitkä globiinigeenit ovat mutaantuneet (esimerkiksi alfa- ja beetageenit), ja sairauden vaikeusasteen mukaan, joka liittyy siihen, periytyvätkö mutaatiot heterotsygoottisesti vai homotsygoottisesti/yhdistelmäheterotsygoottisesti. Tärkein aikuisten hemoglobiinin muoto on Hb A, joka on kahden alfa- ja kahden beetaglobiiniketjun muodostama tetrameeri. Alfa-talassemiassa alfaglobiinin ilmentyminen on puutteellista, ja vastaavasti beetaglobiiniketjuja on liikaa.
Beta-talassemiassa tämä malli on päinvastainen. Globiiniketjujen puutteessa ja epätasapainossa punasolujen hemoglobiinipitoisuus vähenee, mikä johtaa mikrosytoosiin ja hypokromiaan. Lisäksi ylimääräiset alfa- tai beetaglobiiniketjut muodostavat epävakaita tetrameerejä, jotka aiheuttavat hemolyysiä. Alfa- ja beeta-talassemia erotetaan toisistaan aikuisten hemoglobiinin Hb A2:n, kahden alfa- ja kahden delta-globiiniketjun muodostaman tetrameerin, määrän perusteella. Hb A2:n määrä lisääntyy beeta-talassemiassa, koska beetaglobiinin suhteellinen puute mahdollistaa sen, että hemoglobiiniin voidaan sisällyttää enemmän delta-ketjuja.
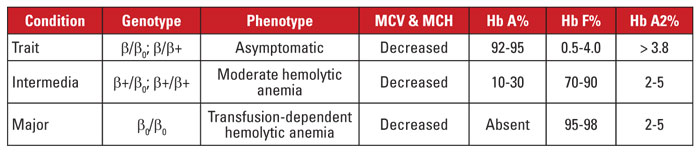
Beta-talassemia johtuu mutaatioista beetaglobiinigeenin geenipaikassa kromosomissa 11.3,4 Suurin osa mutaatioista on pieniä nukleotidisubstituutioita, insertioita tai deleetioita, vaikkakin harvinaisissa tapauksissa on todettu myös suuria deleetioita. Mutaatiosta riippuen beetaglobiinin ilmentyminen vähenee osittain (beetaplus-talassemia) tai kokonaan (beetanolla-talassemia). Beeta-talassemian aiheuttaa heterotsygoottinen mutaatio. Tämä tila on oireeton, ja sille on ominaista lisääntynyt Hb A2, punasolujen mikrosytoosi eikä merkittävää hemolyyttistä anemiaa. Sitä vastoin beeta-talassemia major (Cooleyn anemia) johtuu homotsygoottisesta beeta-nollamutaatiosta. Hemoglobiiniarviossa Hb F on vallitseva, Hb A puuttuu ja Hb A2 on normaali tai lisääntynyt. Beta-talassemia majorille on ominaista vakava, verensiirroista riippuvainen hemolyyttinen anemia, johon liittyy splenomegaliaa ja luuston epämuodostumia. Toistuvat verensiirrot johtavat raudan ylikuormitukseen, joka on tärkein sairastuvuuden ja kuolleisuuden syy. Beta-talassemia intermedia on kliinisesti heterogeeninen, ja useimmat oireet liittyvät keskivaikeaan hemolyyttiseen anemiaan. Verensiirtoriippuvuus on harvinaista, koska beetaglobiinin ilmentyminen ei puutu. Koska beetatalassemiaan liittyy suuri määrä mutaatioita, geneettinen diagnoosi edellyttää yleensä geenien sekvensointia. Taulukossa 3 on yhteenveto beeta-talassemian oireyhtymistä.
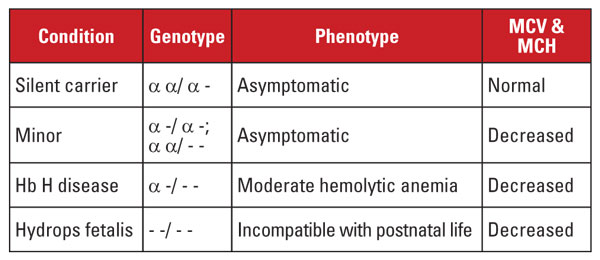
Alfa-talassemia johtuu mutaatioista kromosomissa 16 sijaitsevassa alfaglobiinigeenin lokuksessa.5 Alfaglobiinigeenit ovat kahdentuneet kromosomissa 16, joten normaalilla yksilöllä on neljä kopiota. Yleisimmät mutaatiot ovat suuria deleetioita, jotka vaikuttavat toiseen tai molempiin geeneihin lokuksessa. Deletioita on kuvattu melko rajallinen määrä, ja ne ovat yleisiä eri väestöissä; esimerkiksi suuret mutaatiot, jotka poistavat molemmat alfa-geenit, ovat hyvin yleisiä Kaakkois-Aasian etnisiin ryhmiin kuuluvilla ihmisillä. Näitä mutaatioita esiintyy myös Välimeren alueen väestöissä, mutta ne ovat harvinaisia afrikkalaista alkuperää olevilla ihmisillä. Sitä vastoin yksittäisten alfaglobiinigeenien deletioilla on laaja levinneisyys. Yhden neljästä alfaglobiinigeenistä häviämistä kutsutaan hiljaiseksi kantajatilaksi.
Tämä tila on oireeton, ja siihen liittyy yleensä normaalit punasoluindeksit. Kahden alfaglobiinigeenin menetystä kutsutaan alfatalassemia minoriksi. Tämä tila voi johtua homotsygoottisista yhden geenin deleetioista tai heterotsygoottisista kahden geenin deleetioista. Punasolujen määrä ja indeksit eivät eroa beeta-talassemian ominaisuudesta, mutta Hb A2 -taso on normaali. Kolmen alfageenin menetystä kutsutaan Hb H -taudiksi. (Hb H on beetaglobiiniketjujen tetrameeri.) Hb H -tauti on tyypillisesti keskivaikea hemolyyttinen anemia, johon liittyy splenomegaliaa ja luustomuutoksia. Verensiirtoriippuvuus on harvinaista.
Kaikkien neljän alfageenin menetystä kutsutaan Hb Barts hydrops fetalis -taudiksi (Hb Barts on sikiön gammaglobiiniketjujen tetrameeri). Tämä tila on yhteensopimaton syntymän jälkeisen elämän kanssa. Hb H ja Hb Barts voidaan tunnistaa hemoglobiiniarvioinneissa, ja niillä on merkitystä alfa-talassemian postnataalisessa diagnosoinnissa.5
Geneettinen diagnoosi tehdään yleensä aukkopolymeraasiketjureaktiomäärityksillä (PCR), jotka kohdistuvat yleisiin deleetioihin. Myös alfaglobiinigeenin sekvensointi on käytettävissä harvinaisten mutaatioiden havaitsemiseksi. Taulukossa 4 on yhteenveto alfa-talassemian oireyhtymistä.
Kun geenitestiä tarvitaan
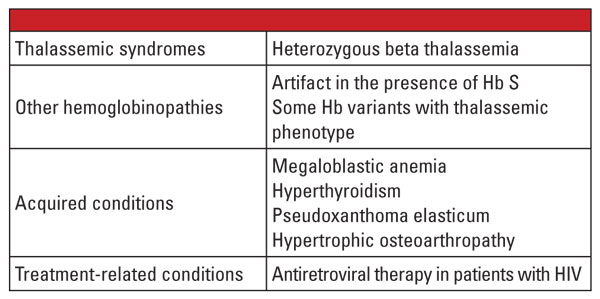
Johtopäätöksenä voidaan todeta, että alfa- ja beeta-talassemian heterotsygoottiset muodot ovat hyvin yleisiä, ja ne tunnistetaan usein kliinisessä laboratoriossa. Hemoglobiinin arviointi ja muut rutiininomaiset kliiniset ja laboratoriotiedot riittävät yleensä diagnoosiin. Geneettinen testaus ei ole useimmissa tapauksissa tarpeen, mutta sillä voi olla ratkaiseva merkitys perhesuunnittelun yhteydessä. Jos molemmilla vanhemmilla todetaan talassemiamutaatioita, sikiöllä voi olla merkittävä riski periä vaikea talassemia. Jos kyseessä on hydrops fetalis, sikiön kuolema on todennäköinen, ja äidille aiheutuu merkittävä riski raskauskomplikaatioista.5 Siksi on tärkeää olla tietoinen muuttujista, jotka vaikeuttavat talassemioiden laboratoriodiagnoosia, ja tunnistaa, milloin varmistava geenitesti on aiheellinen. Esimerkiksi, kuten ensimmäisessä tapauskertomuksessa osoitettiin, beeta-talassemian diagnoosi voi olla haastavaa, jos on olemassa samanaikaisia sairauksia, jotka vaikuttavat Hb A2:n tasoon. Taulukossa 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biologia, kliininen merkitys ja mahdollinen kohde sirppisolusairauden parantamiseksi. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-taalassemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-talassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Geenitestikatsaus. Alfa-talassemia. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH recommendations for the measurement of hemoglobin A2. Int J Lab Hematol. 2012;34(1):1-13.