V běžné laboratorní praxi se diagnóza rysu beta talasemie obvykle stanovuje na základě charakteristických nálezů při hodnocení hemoglobinu a počtu a indexů červených krvinek. Zejména je zvýšené procento hemoglobinu (Hb) A2, zatímco střední korpuskulární objem červených krvinek (MCV) a/nebo střední korpuskulární hemoglobin (MCH) jsou snížené. Počet červených krvinek je obvykle normální nebo zvýšený, ale může být snížený, pokud má pacient jiné příčiny anémie.
Talasemie beta trait (nazývaná také beta talasemie minor nebo stav nosičství beta talasemie) je benigní, heterozygotní stav, který lze podle klinických a laboratorních znaků odlišit od závažnějších syndromů beta talasemie (intermedia a major). Beta talasemie intermedia a major jsou spojeny s narůstající závažností anémie, závislostí na transfuzích a splenomegalií, zatímco u beta talasemie trait tyto znaky chybí. U těžké beta talasemie je hladina fetálního hemoglobinu (Hb F) výrazně zvýšena v důsledku absence Hb A a množství Hb A2 nemusí být zvýšeno jako u beta talasemie trait. Laboratorní diagnostika rysu beta talasemie by tedy měla být poměrně jednoduchá, je to tak?“
Ne vždy. Následující dva případy ilustrují běžné situace, které mohou diagnózu komplikovat.
Případ 1: konkurenční podmínky
Pacientkou byla 28letá Afroameričanka, která byla pozitivní na infekci HIV-1 a podstupovala vysoce aktivní antiretrovirovou léčbu (HAART) s režimem dolutegravir/abakavir/lamivudin. Laboratorní nálezy jsou uvedeny v tabulce 1.
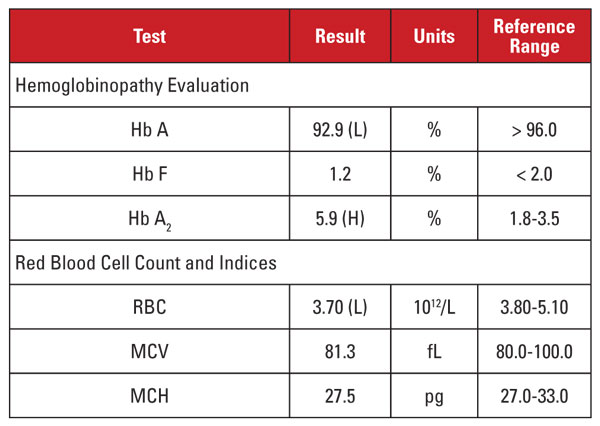
Tento případ představoval zajímavou situaci. Ačkoli byla hladina Hb A2 výrazně zvýšená, odpovídající hemogram neprokazoval mikrocytózu ani hypochromii. Pokud jsou laboratorní znaky diskordantní pro diagnózu beta talasemického rysu, je užitečné zvážit další stavy, které ovlivňují hladinu Hb A2 a indexy červených krvinek. V tomto případě bylo důležité vodítko nalezeno v klinické anamnéze pacienta.
Hladina Hb A2 má tendenci se zvyšovat při stavech, které zpožďují jaderné zrání prekurzorů červených krvinek. Tyto stavy jsou také spojeny se zvýšením MCV.1,2 Nejčastější příčinou tohoto jevu je megaloblastická anémie způsobená nedostatkem folátu a/nebo vitaminu B12. Podobný účinek však mají i některé léky, které inhibují syntézu nukleových kyselin, včetně skupiny léků proti HIV nazývané nukleosidové inhibitory reverzní transkriptázy (NRTI). Lamivudin v režimu HAART tohoto pacienta patří mezi léky NRTI.
Měli bychom tedy dojít k závěru, že vysoká hladina Hb A2 u tohoto pacienta byla způsobena léky proti HIV a pacient neměl rysy beta talasemie? Ne tak rychle! Obecně platí, že zvýšení Hb A2 způsobené NRTI (nebo megaloblastickou anémií) je menší než zvýšení pozorované u rysů beta talasemie.1,2 Navíc toto zvýšení bývá úměrné celkovému účinku léku, který lze přibližně vyjádřit zvýšením MCV.1,2 U mnoha pacientů léčených NRTI je MCV výrazně vysoká (často vyšší než 120 fL), zatímco u našeho pacienta se MCV pohybovala směrem k dolní hranici referenčního rozmezí.
Nebyli jsme proto přesvědčeni, že velmi vysoká hladina Hb A2 u pacienta byla způsobena pouze léčbou lamivudinem. Měli jsme podezření, že pacientka měla konkurenční podmínky, které zvyšovaly a snižovaly MCV (tj. rys beta talasemie, respektive terapie lamivudinem), přičemž obě přispívaly k vysoké hladině Hb A2. Objednávajícímu lékaři jsme sdělili, že rysy beta talasemie jsou pravděpodobné a mohly by být potvrzeny analýzou mutace beta globinu. Následné sekvenování genu odhalilo heterozygotní mutaci beta globinu spojenou s beta-talasemií.
Případ 2: faktor etnicity
Pacientkou byla 35letá těhotná Vietnamka, která byla vyšetřována na hemoglobinopatii. Laboratorní nálezy jsou uvedeny v tabulce 2.
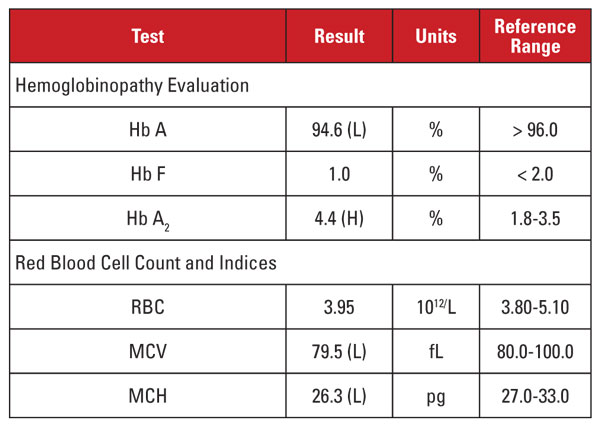
Vysoká hladina Hb A2 byla opět diskordantní s minimální nebo chybějící mikrocytózou a hypochromií. V tomto případě jsme mohli vyloučit běžné matoucí faktory, jako je nedostatek folátů, vitaminu B12 nebo železa, dysfunkce štítné žlázy nebo účinky léků. Na základě etnického původu pacienta jsme pojali podezření na další důležitou příčinu diskordance: znak beta talasemie děděný společně s alfa talasemií minor. Pokud pacient zdědí oba tyto stavy, MCV a MCH se často normalizují, protože alfa a beta globinové řetězce jsou ve vyvíjejících se červených krvinkách přítomny v relativně vyváženém množství.2
Doporučili jsme genetické analýzy k vyšetření mutací alfa a beta talasemie, aby bylo možné plně posoudit riziko pro těhotnou matku a plod. Následné sekvenování beta globinu odhalilo heterozygotní mutaci beta plus talasemie, zatímco analýza delece alfa globinu odhalila heterozygotní deleci dvou genů jihovýchodní Asie (SEA), odpovídající -/αα alfa talasemii minor. Na základě těchto nálezů byla indikována genetická analýza otce k posouzení rizika pro plod, že zdědí klinicky závažný talasemický syndrom.
Talasemie alfa a beta
Talasemie patří mezi nejčastější dědičné poruchy u lidstva. Jsou velmi rozšířené tam, kde se endemicky vyskytovala malárie, a v důsledku migrace lidské populace jsou nyní běžné ve všech částech světa. Talasemie jsou způsobeny mutacemi, které snižují expresi globinových genů v prekurzorech červených krvinek. Poruchy se dělí podle toho, které globinové geny jsou mutovány (například alfa versus beta), a podle závažnosti onemocnění, která souvisí s tím, zda se mutace dědí heterozygotně nebo homozygotně/složeně heterozygotně. Hlavní formou hemoglobinu u dospělých je Hb A, tetramer dvou globinových řetězců alfa a dvou beta. U talasemie alfa je exprese globinu alfa nedostatečná a je zde odpovídající nadbytek řetězců globinu beta.
Tento vzorec je u talasemie beta obrácený. Při nedostatku a nerovnováze globinových řetězců je obsah hemoglobinu v červených krvinkách snížený, což vede k mikrocytóze a hypochromii. Nadbytek alfa nebo beta globinových řetězců navíc tvoří nestabilní tetramery, které způsobují hemolýzu. Alfa a beta talasemie se rozlišují podle množství minoritního dospělého hemoglobinu Hb A2, tetrameru dvou alfa a dvou delta globinových řetězců. Hb A2 je u beta-talasemie zvýšený, protože relativní nedostatek beta-globinu umožňuje začlenit do hemoglobinu více delta-řetězců.
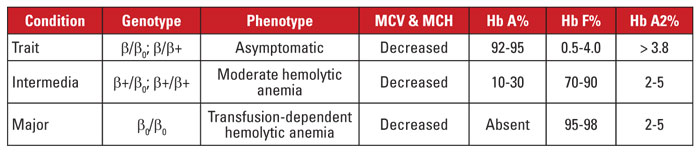
Beta-talasemie je způsobena mutacemi v lokusu beta-globinového genu na chromozomu 11.3,4 Většina mutací jsou malé nukleotidové substituce, inzerce nebo delece, i když ve vzácných případech jsou identifikovány velké delece. V závislosti na mutaci je exprese beta globinu snížena částečně (beta plus talasemie) nebo zcela (beta nulová talasemie). Znak beta talasemie je způsoben heterozygotní mutací. Tento stav je asymptomatický a je charakterizován zvýšeným Hb A2, mikrocytózou červených krvinek a žádnou významnou hemolytickou anémií. Naproti tomu beta talasemie major (Cooleyova anémie) je způsobena homozygotní mutací beta nula. Při hodnocení hemoglobinu se zjistí převaha Hb F, chybějící Hb A a normální nebo zvýšený Hb A2. Beta talasemie major je charakterizována těžkou, na transfuzích závislou hemolytickou anémií s následnou splenomegalií a kostními deformitami. Opakované transfuze vedou k přetížení železem, které je hlavní příčinou morbidity a mortality. Beta talasemie intermedia je klinicky heterogenní, většina příznaků souvisí se středně těžkou hemolytickou anémií. Závislost na transfuzi je neobvyklá, protože exprese beta globinu nechybí. Vzhledem k velkému počtu mutací spojených s beta talasemií vyžaduje genetická diagnostika obvykle sekvenování genů. Syndromy beta talasemie jsou shrnuty v tabulce 3.
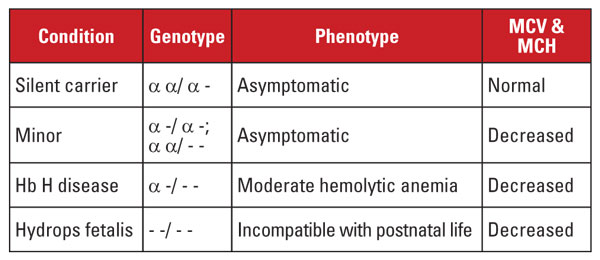
Alfa talasemie je způsobena mutacemi v lokusu alfa globinového genu na chromozomu 16.5 Alfa globinové geny jsou na chromozomu 16 duplikovány, takže normální jedinec má čtyři kopie. Nejčastějšími mutacemi jsou velké delece, které postihují jeden nebo oba geny na lokusu. Bylo popsáno poměrně omezené množství delecí s výskytem v různých populacích; například velké mutace, které odstraňují oba alfa geny, jsou velmi časté u lidí z etnika jihovýchodní Asie. Tyto mutace jsou také časté u středomořské populace, ale jsou vzácné u lidí afrického etnika. Naproti tomu delece jednotlivých alfa globinových genů mají široké rozšíření. Ztráta jednoho ze čtyř alfa globinových genů se označuje jako stav tichého přenašeče.
Tento stav je asymptomatický a obvykle je spojen s normálními ukazateli červených krvinek. Ztráta dvou alfa globinových genů se označuje jako alfa talasemie minor. Tento stav může nastat v důsledku homozygotní delece jednoho genu nebo heterozygotní delece dvou genů. Počet a indexy červených krvinek jsou k nerozeznání od rysu beta talasemie, ale hladina Hb A2 je normální. Ztráta tří genů alfa se označuje jako nemoc Hb H. (Hb H je tetramer beta globinových řetězců.) Pro nemoc Hb H je typická středně těžká hemolytická anémie spojená se splenomegalií a kostními změnami. Závislost na transfuzi je neobvyklá.
Ztráta všech čtyř alfa genů se označuje jako Hb Barts hydrops fetalis (Hb Barts je tetramer fetálních gama globinových řetězců). Tento stav je neslučitelný s postnatálním životem. Hb H a Hb Barts lze identifikovat při hodnocení hemoglobinu a hrají roli při postnatální diagnostice alfa talasemie.5
Genetická diagnostika se obvykle provádí pomocí testů s mezerami polymerázové řetězové reakce (PCR), které se zaměřují na běžné delece. K detekci vzácných mutací je k dispozici také sekvenování alfa globinového genu. Syndromy alfa talasemie jsou shrnuty v tabulce 4.
Kdy je nutné genetické vyšetření
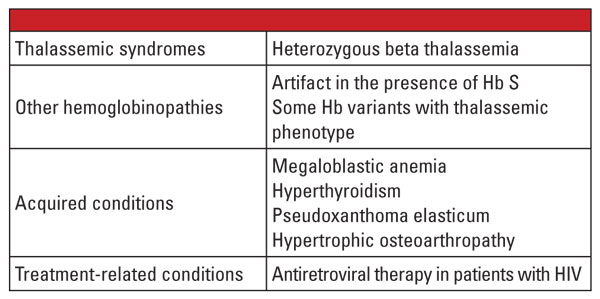
Závěrem lze říci, že heterozygotní formy alfa a beta talasemie jsou velmi časté a jsou často identifikovány v klinické laboratoři. Vyhodnocení hemoglobinu a další běžné klinické a laboratorní údaje jsou obvykle pro stanovení diagnózy dostačující. Genetické vyšetření není ve většině případů nutné, ale může být kriticky důležité v souvislosti s plánováním rodiny. Pokud jsou mutace talasemie identifikovány u obou rodičů, může existovat významné riziko pro plod, že zdědí těžkou talasemii. V případě hydrops fetalis je pravděpodobný zánik plodu a pro matku existuje významné riziko komplikací těhotenství.5 Proto je důležité znát proměnné, které komplikují laboratorní diagnostiku talasemií, a rozpoznat, kdy je indikováno potvrzující genetické vyšetření. Například, jak ukazuje první kazuistika, diagnostika rysu beta talasemie může být náročná, pokud existují souběžné stavy, které ovlivňují hladinu Hb A2. Faktory, o kterých je známo, že zvyšují hladinu Hb A2, jsou uvedeny v tabulce 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biologie, klinický význam a možný cíl pro zlepšení srpkovité choroby. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-thalasemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Přehled genových testů. Alfa-talasemie. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. Doporučení ICSH pro měření hemoglobinu A2. Int J Lab Hematol. 2012;34(1):1-13.