I rutinmässig laboratoriepraxis ställs diagnosen beta thalassemia trait vanligen genom karakteristiska fynd i hemoglobinutvärderingen och antalet röda blodkroppar och index. I synnerhet är andelen hemoglobin (Hb) A2 förhöjd, medan de röda blodkropparnas medelkroppsvolym (MCV) och/eller medelkroppshemoglobin (MCH) är nedsatta. Antalet röda blodkroppar är vanligtvis normalt eller förhöjt, men kan vara sänkt om patienten har andra orsaker till anemi.
Beta thalassemia trait (även kallat beta thalassemia minor eller beta thalassemia carrier state) är ett godartat, heterozygot tillstånd som kan särskiljas från de allvarligare beta thalassemia-syndromen (intermedia och major) med hjälp av kliniska och laboratorieegenskaper. Betathalassemi intermedia och major är förknippade med ökande svårighetsgrad av anemi, transfusionsberoende och splenomegali, medan dessa egenskaper saknas vid betathalassemi trait. Vid svår betathalassemi är nivån av fetalt hemoglobin (Hb F) markant förhöjd på grund av avsaknaden av Hb A, och mängden Hb A2 kanske inte är förhöjd som vid betathalassemi trait. Så laboratoriediagnosen för beta thalassemia trait borde vara relativt enkel, eller hur?
Inte alltid. Följande två fall illustrerar vanliga situationer som kan komplicera diagnosen.
Fall 1: konkurrerande förhållanden
Patienten var en 28-årig afroamerikansk kvinna som var positiv för hiv-1-infektion och genomgick högaktiv antiretroviral terapi (HAART) med en dolutegravir/abacavir/lamivudin-regim. Laboratoriefynd visas i tabell 1.
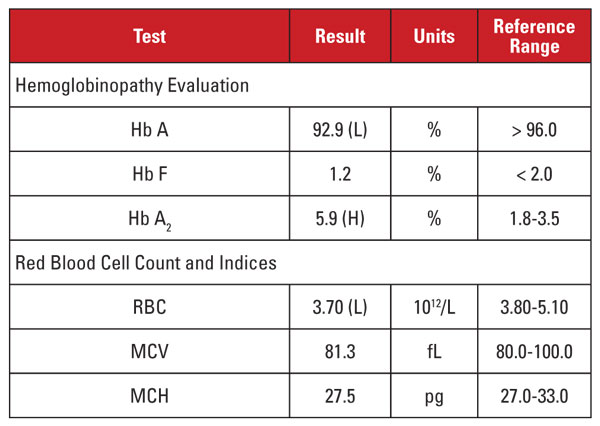
Detta fall presenterade en intressant situation. Även om Hb A2-nivån var betydligt förhöjd visade motsvarande hemogram inte på mikrocytos eller hypokromi. När laboratorieegenskaperna inte stämmer överens med diagnosen beta thalassemia trait är det lämpligt att överväga andra tillstånd som påverkar Hb A2-nivån och index för röda blodkroppar. I det här fallet hittades en viktig ledtråd i patientens kliniska historia.
Hb A2-nivåerna tenderar att öka i tillstånd som fördröjer kärnmognaden hos prekursorer av röda blodkroppar. Dessa tillstånd är också förknippade med ökat MCV.1,2 Den vanligaste orsaken till detta fenomen är megaloblastisk anemi på grund av folat- och/eller vitamin B12-brist. Flera läkemedel som hämmar nukleinsyrasyntesen har dock en liknande effekt, inklusive klassen av anti-HIV-läkemedel som kallas nukleosidhämmare av omvänt transkriptas (NRTI). Lamivudin i den här patientens HAART-regim är ett NRTI-läkemedel.
Så ska vi dra slutsatsen att den här patientens höga Hb A2-nivå berodde på anti-HIV-läkemedlet och att patienten inte hade beta-thalassemiegenskap? Inte så snabbt! I allmänhet är ökningar av Hb A2 på grund av NRTI (eller megaloblastisk anemi) mindre än vad som ses vid beta thalassemi trait.1,2 Dessutom tenderar ökningarna att vara proportionella mot läkemedlets övergripande effekter, vilket kan approximeras genom ökningar av MCV.1,2 Hos många patienter som får NRTI-behandling är MCV tydligt högt (ofta större än 120 fL), medan MCV hos vår patient låg mot den nedre delen av referensintervallet.
Därmed var vi inte övertygade om att patientens mycket höga Hb A2-nivå enbart berodde på lamivudinbehandling. Vi misstänkte att patienten hade konkurrerande tillstånd som ökade respektive minskade MCV (dvs. beta-thalassemiegenskap respektive lamivudinbehandling), där båda bidrog till en hög Hb A2-nivå. Vi rapporterade till den beställande läkaren att beta-thalassemi var troligt och att det kunde bekräftas genom analys av betaglobinmutationer. Efterföljande gensekvensering avslöjade en heterozygot betaglobinmutation associerad med beta-zero thalassemi.
Fall 2: etnicitetsfaktor
Patienten var en 35-årig gravid vietnamesisk kvinna som undersöktes för hemoglobinopati. Laboratoriefynd visas i tabell 2.
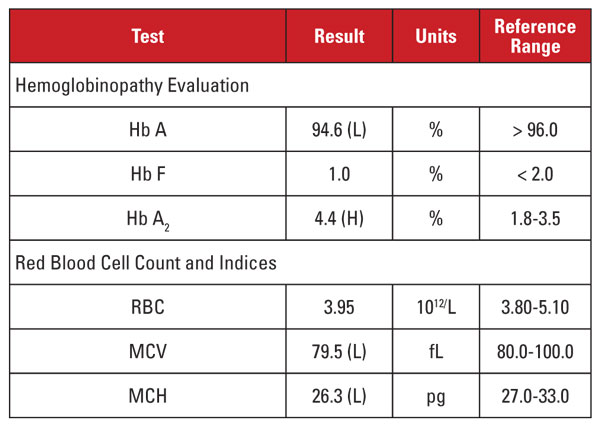
Den höga Hb A2-nivån var återigen diskordant med minimal eller frånvarande mikrocytos och hypokromi. I det här fallet kunde vi utesluta vanliga förväxlingsfaktorer som folat-, vitamin B12- eller järnbrist, sköldkörteldysfunktion eller medicineringseffekter. Baserat på patientens etnicitet misstänkte vi en annan viktig orsak till diskordansen: beta thalassemia trait som är medfödd med alfa thalassemia minor. När en patient ärver båda dessa tillstånd normaliseras ofta MCV och MCH eftersom alfa- och betaglobinkedjor finns i relativt balanserade mängder i röda blodkroppar under utveckling.2
Vi rekommenderade genetiska analyser för att testa alfa- och betathalassemimutationer, för att fullt ut kunna bedöma risken för den gravida modern och fostret. Efterföljande sekvensering av betaglobin avslöjade en heterozygot beta-plus thalassemiamutation, medan analysen av alfaglobindeletioner avslöjade en heterozygot sydostasiatisk (SEA) deletion med två gener, vilket stämmer överens med -/ααα alfa thalassemi minor. Baserat på dessa fynd var genetisk analys av fadern indicerad för att bedöma risken för fostret att ärva ett kliniskt allvarligt thalassemisyndrom.
Alpha- och betathalassemier
Thalassemier är bland de vanligaste ärftliga sjukdomarna i mänskligheten. De är mycket vanliga där malaria har varit endemisk och är nu vanliga i alla delar av världen på grund av migration av mänskliga populationer. Thalassemier orsakas av mutationer som minskar globin-genuttrycket i prekursorer av röda blodkroppar. Sjukdomarna klassificeras efter vilka globingener som är muterade (t.ex. alfa- eller betagener) och efter sjukdomens svårighetsgrad, som är relaterad till om mutationerna ärvs på ett heterozygot eller homozygot/komplett heterozygot sätt. Den viktigaste formen av hemoglobin hos vuxna är Hb A, en tetramer av två alfa- och två betaglobinkedjor. Vid alfa-thalassemi är alfaglobinuttrycket bristfälligt och det finns ett motsvarande överskott av betaglobinkedjor.
Detta mönster är omvänt vid betatalassemi. Vid brist och obalans i globinkedjorna minskar hemoglobinhalten i de röda blodkropparna, vilket resulterar i mikrocytos och hypokromi. Dessutom bildar överskott av alfa- eller betaglobinkedjor instabila tetramerer som orsakar hemolys. Alfa- och betathalassemi skiljer sig åt genom mängden av det mindre vuxna hemoglobinet Hb A2, en tetramer av två alfa- och två deltaglobinkedjor. Hb A2 ökar vid betathalassemi eftersom den relativa bristen på betaglobin gör att fler deltakedjor kan inkorporeras i hemoglobin.
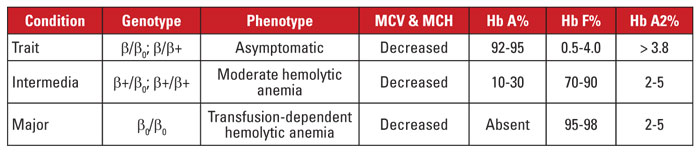
Betathalassemi orsakas av mutationer i betaglobingenen locus på kromosom 11.3,4 De flesta mutationer är små nukleotidssubstitutioner, insättningar eller deletioner, även om stora deletioner identifieras i sällsynta fall. Beroende på mutationen reduceras betaglobinuttrycket delvis (beta-plus thalassemi) eller helt (beta-zero thalassemi). Betathalassemiegenskapen orsakas av en heterozygot mutation. Detta tillstånd är asymtomatiskt och kännetecknas av ökad Hb A2, mikrocytos i de röda blodkropparna och ingen betydande hemolytisk anemi. Betathalassemi major (Cooleys anemi) orsakas däremot av homozygota beta-zero-mutationer. Hemoglobinutvärderingen visar en dominans av Hb F, avsaknad av Hb A och normalt eller förhöjt Hb A2. Beta thalassemi major kännetecknas av allvarlig, transfusionsberoende hemolytisk anemi med splenomegali och benmissbildningar som följd. Återkommande transfusioner leder till järnöverbelastning, vilket är en huvudorsak till sjuklighet och dödlighet. Beta thalassemia intermedia är kliniskt heterogen, med de flesta symtom relaterade till måttlig hemolytisk anemi. Transfusionsberoende är ovanligt eftersom betaglobinuttryck inte saknas. På grund av det stora antalet mutationer som är förknippade med betathalassemi kräver genetisk diagnos vanligtvis gensekvensering. Betathalassemiasyndromen sammanfattas i tabell 3.
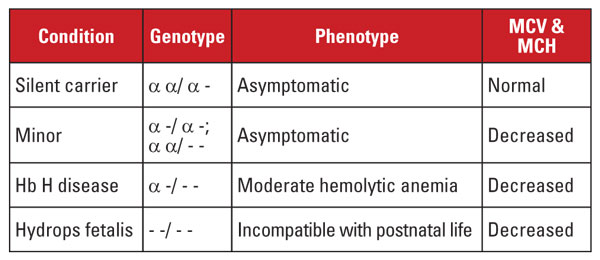
Alpha thalassemi orsakas av mutationer i alfaglobingenens locus på kromosom 16.5 Alfaglobingenerna är duplicerade på kromosom 16, så en normal individ har fyra kopior. De vanligaste mutationerna är stora deletioner som påverkar en eller båda generna på lokus. Ett ganska begränsat antal deletioner har beskrivits, med prevalens i olika populationer; till exempel är stora mutationer som raderar båda alfa-generna mycket vanliga hos personer av sydostasiatisk etnicitet. Dessa mutationer är också vanliga i Medelhavspopulationer, men är sällsynta hos personer av afrikansk etnicitet. Däremot har deletioner av enskilda alfa-globingener en utbredd spridning. Förlusten av en av de fyra alfaglobingenerna kallas ett tyst bärartillstånd.
Detta tillstånd är asymtomatiskt och är vanligen förknippat med normala index för röda blodkroppar. Förlusten av två alfaglobingener kallas alfa-thalassemi minor. Detta tillstånd kan uppstå på grund av homozygota deletioner av en gen eller heterozygota deletioner av två gener. Antalet röda blodkroppar och index skiljer sig inte från beta thalassemia trait, men Hb A2-nivån är normal. Förlusten av tre alfagener kallas Hb H-sjukdom. (Hb H är en tetramer av betaglobinkedjor.) Hb H-sjukdom är typiskt en måttlig hemolytisk anemi i samband med splenomegali och benförändringar. Transfusionsberoende är ovanligt.
Förlust av alla fyra alfagener kallas Hb Barts hydrops fetalis (Hb Barts är en tetramer av fetala gammaglobinkedjor). Detta tillstånd är oförenligt med livet efter födseln. Hb H och Hb Barts kan identifieras i hemoglobinutvärderingar och spelar en roll vid postnatal diagnos av alfa-thalassemi.5
Den genetiska diagnosen utförs vanligen med hjälp av gap-polymerasekedjereaktionsanalyser (PCR) som är inriktade på de vanliga deletionerna. Sekvensering av alfaglobingenen är också tillgänglig för att upptäcka sällsynta mutationer. Alfa-thalassemiasyndromen sammanfattas i tabell 4.
När genetisk testning behövs
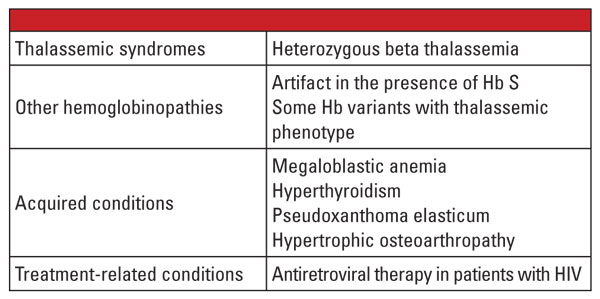
Slutsatsen är att de heterozygota formerna av alfa- och betathalassemi är mycket vanliga och identifieras ofta i det kliniska laboratoriet. Hämoglobinutvärdering och andra kliniska rutin- och laboratoriedata är vanligtvis tillräckliga för diagnosen. Genetisk testning är inte nödvändig i de flesta fall, men kan vara av avgörande betydelse i samband med familjeplanering. Om thalassemimutationer identifieras hos båda föräldrarna kan det finnas en betydande risk för fostret att ärva svår thalassemi. Vid hydrops fetalis är det troligt att fostret dör och det finns en betydande risk för modern att drabbas av graviditetskomplikationer.5 Därför är det viktigt att vara medveten om variabler som komplicerar laboratoriediagnosen av thalassemier och att känna igen när bekräftande genetisk testning är indicerad. Som framgår av den första fallrapporten kan det till exempel vara svårt att ställa diagnosen beta-thalassemiatrait om det finns samexisterande tillstånd som påverkar nivån av Hb A2. Faktorer som är kända för att öka Hb A2 anges i tabell 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biologi, klinisk relevans och ett möjligt mål för att förbättra sicklecellsjukdom. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-thalassemi. Genet Med. 2010;12(2):61-76.
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Gene test review. Alpha-thalassemi. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH-rekommendationer för mätning av hemoglobin A2. Int J Lab Hematol. 2012;34(1):1-13.