În practica de laborator de rutină, diagnosticul de trăsătură beta talasemică este de obicei pus prin constatări caracteristice în evaluarea hemoglobinei și a numărului și indicilor de globule roșii. În special, procentul de hemoglobină (Hb) A2 este ridicat, în timp ce volumul corpuscular mediu al globulelor roșii (MCV) și/sau hemoglobina corpusculară medie (MCH) sunt scăzute. Numărul de globule roșii este de obicei normal sau crescut, dar poate fi scăzut dacă pacientul are alte cauze de anemie.
Trasatura beta talasemică (numită și beta talasemie minoră sau stare de purtător de beta talasemie) este o afecțiune benignă, heterozigotă, care poate fi distinsă de sindroamele beta talasemice mai severe (intermedia și majoră) prin caracteristicile clinice și de laborator. Beta talasemia intermedia și majoră sunt asociate cu o severitate din ce în ce mai mare a anemiei, dependență de transfuzii și splenomegalie, în timp ce aceste caracteristici sunt absente în cazul trăsăturii beta talasemiei. În beta talasemia severă, nivelul de hemoglobină fetală (Hb F) este marcat de o creștere accentuată din cauza absenței Hb A, iar cantitatea de Hb A2 poate să nu fie crescută ca în cazul trăsăturii beta talasemice. Așadar, diagnosticul de laborator al trăsăturii beta talasemiei ar trebui să fie relativ simplu, corect?
Nu întotdeauna. Următoarele două cazuri ilustrează situații comune care pot complica diagnosticul.
Cazul 1: condiții concurente
Pacienta era o femeie afro-americană în vârstă de 28 de ani care era pozitivă la infecția cu HIV-1 și urma un tratament antiretroviral foarte activ (HAART) cu un regim dolutegravir/abacavir/lamivudină. Rezultatele de laborator sunt prezentate în tabelul 1.
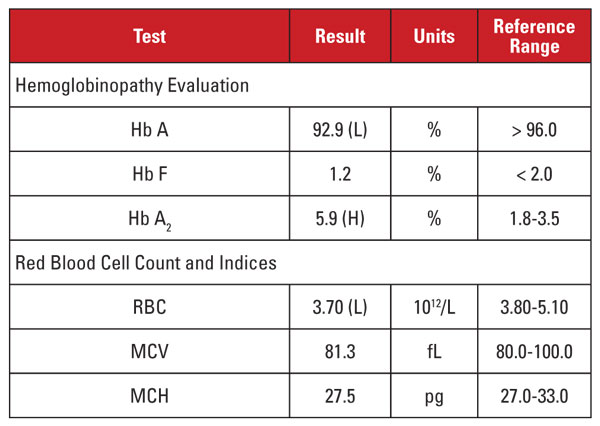
Acest caz a prezentat o situație interesantă. Deși nivelul Hb A2 a fost semnificativ ridicat, hemograma corespunzătoare nu a demonstrat microcitoză sau hipocromie. Atunci când caracteristicile de laborator sunt discordante pentru un diagnostic de trăsătură beta talasemică, este util să se ia în considerare alte afecțiuni care afectează nivelul Hb A2 și indicii globulelor roșii. În acest caz, un indiciu important a fost găsit în istoricul clinic al pacientului.
Nivelul de Hb A2 tinde să crească în condițiile care întârzie maturarea nucleară a precursorilor globulelor roșii. Aceste afecțiuni sunt, de asemenea, asociate cu creșterea MCV.1,2 Cea mai frecventă cauză a acestui fenomen este anemia megaloblastică datorată deficienței de folat și/sau vitamina B12. Cu toate acestea, mai multe medicamente care inhibă sinteza acizilor nucleici au un efect similar, inclusiv clasa de medicamente anti-HIV numită inhibitori nucleozidici ai transcriptazei inverse (NRTI). Lamivudina din regimul HAART al acestui pacient este un medicament NRTI.
Deci ar trebui să concluzionăm că nivelul ridicat de Hb A2 al acestui pacient s-a datorat medicației anti-HIV, iar pacientul nu avea trăsătură beta talasemică? Nu atât de repede! În general, creșterile de Hb A2 datorate NRTI (sau anemiei megaloblastice) sunt mai mici decât cele observate în cazul trăsăturii beta talasemice.1,2 În plus, creșterile tind să fie proporționale cu efectele generale ale medicamentului, care pot fi aproximate prin creșteri ale MCV.1,2 La mulți pacienți care urmează un tratament cu NRTI, MCV-ul este marcant de ridicat (adesea mai mare de 120 fL), în timp ce la pacientul nostru, MCV-ul era spre capătul inferior al intervalului de referință.
De aceea, nu am fost convinși că nivelul foarte ridicat de Hb A2 al pacientului s-a datorat exclusiv tratamentului cu lamivudină. Am suspectat că pacientul a avut condiții concurente care au crescut și au scăzut MCV (adică trăsătura beta talasemică și, respectiv, terapia cu lamivudină), ambele contribuind la un nivel ridicat de Hb A2. I-am raportat medicului curant că trăsătura beta talasemică era probabilă și putea fi confirmată prin analiza mutației beta globinei. Secvențierea genetică ulterioară a evidențiat o mutație heterozigotă a beta globinei asociată cu talasemia beta zero.
Cazul 2: factor etnic
Pacienta era o femeie vietnameză însărcinată în vârstă de 35 de ani, care a fost supusă unui screening pentru hemoglobinopatie. Rezultatele de laborator sunt prezentate în tabelul 2.
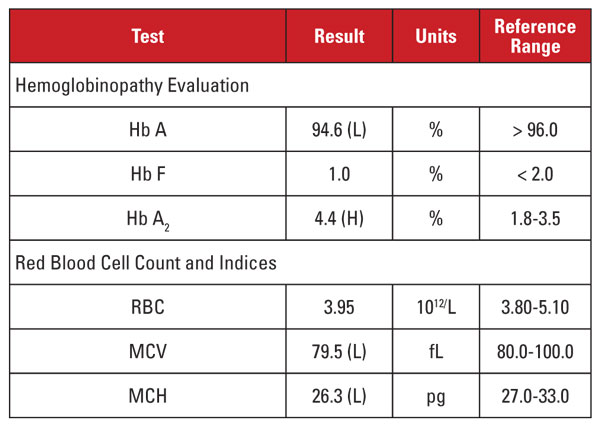
Din nou, nivelul ridicat de Hb A2 a fost în discordanță cu microcitoza și hipocromia minime sau absente. În acest caz, am reușit să excludem factorii de confuzie comuni, cum ar fi deficiența de folat, vitamina B12 sau fier, disfuncția tiroidiană sau efectele medicamentelor. Pe baza apartenenței etnice a pacientului, am suspectat o altă cauză importantă de discordanță: trăsătura beta talasemică coeredată cu alfa talasemia minoră. Atunci când un pacient moștenește ambele afecțiuni, MCV și MCH se normalizează adesea, deoarece lanțurile de alfa și beta globină sunt prezente în cantități relativ echilibrate în globulele roșii în curs de dezvoltare.2
Am recomandat analize genetice pentru a testa mutațiile alfa și beta talasemiei, pentru a evalua pe deplin riscul pentru mama însărcinată și pentru făt. Secvențierea ulterioară a beta globinei a evidențiat o mutație beta-plus talasemică heterozigotă, în timp ce analiza pentru delețiile alfa globinei a evidențiat o deleție heterozigotă din Asia de Sud-Est (SEA) cu două gene, în concordanță cu -/αα alfa talasemie minoră. Pe baza acestor constatări, analiza genetică a tatălui a fost indicată pentru a evalua riscul ca fătul să moștenească un sindrom talasemic clinic sever.
Talasemiile alfa și beta
Talasemiile se numără printre cele mai frecvente afecțiuni moștenite în omenire. Ele sunt foarte răspândite acolo unde malaria a fost endemică, iar în prezent sunt frecvente în orice parte a lumii datorită migrației populațiilor umane. Talasemiile sunt cauzate de mutații care reduc expresia genei globinei în precursorii globulelor roșii. Tulburările sunt clasificate în funcție de genele globinei care sunt mutate (de exemplu, alfa versus beta) și de gravitatea bolii, care este legată de faptul că mutațiile sunt moștenite în mod heterozigot sau homozigot/heterozigot/compus. Forma majoră de hemoglobină la adult este Hb A, un tetramer format din două lanțuri de globină alfa și două lanțuri de globină beta. În cazul alfa talasemiei, expresia alfa globinei este deficitară și există un exces corespunzător de lanțuri de beta globină.
Acest model este inversat în cazul beta talasemiei. În cadrul deficienței și dezechilibrului lanțurilor globinice, conținutul de hemoglobină din celulele roșii este scăzut, ceea ce duce la microcitoză și hipocromie. În plus, lanțurile de globină alfa sau beta în exces formează tetrameri instabili care provoacă hemoliză. Talasemia alfa și beta se disting prin cantitatea de hemoglobină minoră a adultului Hb A2, un tetramer format din două lanțuri de globină alfa și două lanțuri de globină delta. Hb A2 este crescută în talasemia beta deoarece lipsa relativă de globină beta permite încorporarea mai multor lanțuri delta în hemoglobină.
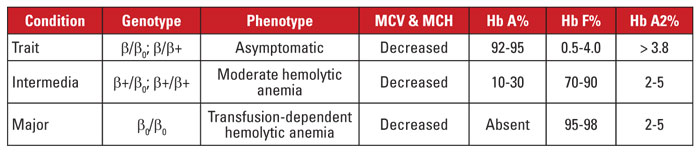
Beta talasemia este cauzată de mutații în locusul genei beta globinei de pe cromozomul 11.3,4 Majoritatea mutațiilor sunt mici substituții, inserții sau deleții de nucleotide, deși în cazuri rare sunt identificate deleții mari. În funcție de mutație, expresia beta globinei este redusă parțial (talasemie beta-plus) sau complet (talasemie beta-zero). Trăsătura beta talasemiei este cauzată de o mutație heterozigotă. Această afecțiune este asimptomatică și se caracterizează prin Hb A2 crescută, microcitoză eritrocitară și fără anemie hemolitică semnificativă. În schimb, beta talasemia majoră (anemia lui Cooley) este cauzată de mutații homozigote beta-zero. Evaluarea hemoglobinei relevă o predominanță a Hb F, Hb A absentă și Hb A2 normală sau crescută. Beta talasemia majoră se caracterizează prin anemie hemolitică severă, dependentă de transfuzii, cu splenomegalie și deformări osoase rezultate. Transfuziile recurente duc la supraîncărcare cu fier, care este o cauză principală de morbiditate și mortalitate. Beta talasemia intermedia este heterogenă din punct de vedere clinic, cu majoritatea simptomelor legate de anemia hemolitică moderată. Dependența de transfuzii este neobișnuită, deoarece expresia beta globinei nu este absentă. Din cauza numărului mare de mutații asociate cu beta talasemia, diagnosticul genetic necesită, de obicei, secvențierea genelor. Sindroamele beta talasemiei sunt rezumate în tabelul 3.
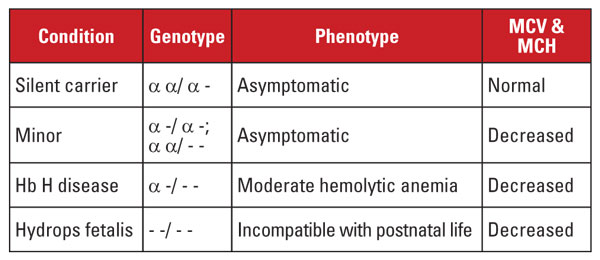
Alfa talasemia este cauzată de mutații în locusul genei alfa globinei de pe cromozomul 16.5 Genele alfa globinei sunt duplicate pe cromozomul 16, astfel încât un individ normal are patru copii. Cele mai frecvente mutații sunt deleții mari care afectează una sau ambele gene de pe locus. A fost descris un număr destul de limitat de deleții, cu prevalență în diferite populații; de exemplu, mutațiile mari care elimină ambele gene alfa sunt foarte frecvente la persoanele de etnie sud-est asiatică. Aceste mutații sunt, de asemenea, prevalente în populațiile mediteraneene, dar sunt rare la persoanele de etnie africană. În schimb, delețiile unor singure gene alfa globinice au o distribuție largă. Pierderea uneia dintre cele patru gene alfa globinice este denumită stare de purtător silențios.
Această afecțiune este asimptomatică și este de obicei asociată cu indici normali ai globulelor roșii. Pierderea a două gene alfa globinice este denumită alfa talasemie minoră. Această afecțiune poate apărea din cauza delețiilor homozigote de o genă sau heterozigote de două gene. Numărul de globule roșii și indicii nu se disting de trăsătura beta talasemiei, dar nivelul Hb A2 este normal. Pierderea a trei gene alfa se numește boala Hb H. (Hb H este un tetramer de lanțuri de beta globină.) Boala Hb H este de obicei o anemie hemolitică moderată asociată cu splenomegalie și modificări osoase. Dependența de transfuzii este neobișnuită.
Pierderea tuturor celor patru gene alfa este denumită Hb Barts hydrops fetalis (Hb Barts este un tetramer de lanțuri de gamaglobină fetală). Această afecțiune este incompatibilă cu viața postnatală. Hb Hb și Hb Barts pot fi identificate în evaluările hemoglobinei și joacă un rol în diagnosticul postnatal al talasemiei alfa.5
Diagnosticul genetic se realizează de obicei prin teste de reacție în lanț a polimerazei în gol (PCR) care vizează delețiile comune. Secvențierea genei alfa globinei este, de asemenea, disponibilă pentru a detecta mutațiile rare. Sindroamele alfa talasemiei sunt rezumate în tabelul 4.
Când este necesară testarea genetică
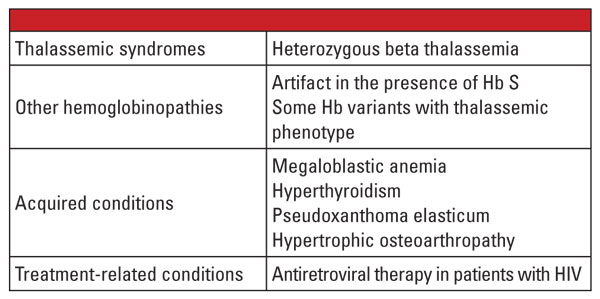
În concluzie, formele heterozigote de alfa și beta talasemie sunt foarte comune și sunt identificate frecvent în laboratorul clinic. Evaluarea hemoglobinei și alte date clinice și de laborator de rutină sunt de obicei suficiente pentru diagnostic. Testarea genetică nu este necesară în majoritatea cazurilor, dar poate avea o importanță critică în contextul planificării familiale. În cazul în care mutațiile talasemiei sunt identificate la ambii părinți, poate exista un risc semnificativ pentru făt de a moșteni talasemia severă. În cazul hidropsului fetal, este probabilă decesul fetal și există un risc semnificativ pentru mamă de complicații ale sarcinii.5 Prin urmare, este important să se cunoască variabilele care complică diagnosticul de laborator al talasemiilor și să se recunoască momentul în care este indicată efectuarea unui test genetic de confirmare. De exemplu, după cum se arată în primul raport de caz, diagnosticul trăsăturii beta talasemiei poate fi dificil dacă există condiții coexistente care afectează nivelul Hb A2. Factorii despre care se știe că măresc Hb A2 sunt enumerați în tabelul 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Disorders of Hemoglobin: Genetics, Pathophysiology Clinical Management. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biologie, relevanță clinică și o posibilă țintă pentru ameliorarea bolii celulelor secerătoare. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-talasemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-Talasemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Gene test review. Alfa-talasemie. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. Recomandări ICSH pentru măsurarea hemoglobinei A2. Int J Lab Hematol. 2012;34(1):1-13.