Na prática laboratorial de rotina, o diagnóstico do traço de talassemia beta é geralmente feito por achados característicos na avaliação da hemoglobina e no hemograma e índices de eritrócitos. Em especial, a percentagem de hemoglobina (Hb) A2 é elevada, enquanto o volume corpuscular médio (VGM) e/ou a hemoglobina corpuscular média (HGM) são diminuídos. A contagem de hemácias em geral é normal ou aumentada, mas pode diminuir se o paciente tem outras causas de anemia.
Beta thalassemia trait (também chamada beta talassemia minor ou beta talassemia carrier state) é uma condição benigna, heterozigótica, que pode ser distinguida das síndromes de beta talassemia mais graves (intermedia e major) por características clínicas e laboratoriais. As talassemias beta intermedia e major estão associadas com o aumento da gravidade da anemia, dependência transfusional e esplenomegalia, enquanto estas características estão ausentes no traço de talassemia beta. Na talassemia beta grave, o nível de hemoglobina fetal (Hb F) está acentuadamente aumentado devido à ausência de Hb A, e a quantidade de Hb A2 pode não estar aumentada como no traço de talassemia beta. Portanto, o diagnóstico laboratorial do traço de talassemia beta deve ser relativamente simples, correto?
Nem sempre. Os dois casos seguintes ilustram situações comuns que podem complicar o diagnóstico.
Caso 1: condições de competição
A paciente era uma afro-americana de 28 anos de idade que era positiva para a infecção pelo HIV-1 e que estava sendo submetida a terapia anti-retroviral altamente ativa (HAART) com regime dolutegravir/abacavir/lamivudina. Os resultados laboratoriais são apresentados na Tabela 1.
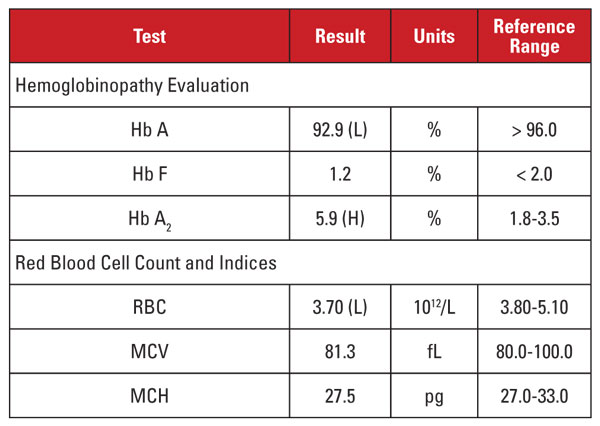
Este caso apresentou uma situação interessante. Embora o nível de Hb A2 tenha sido significativamente elevado, o hemograma correspondente não demonstrou microcitose ou hipocromia. Quando as características laboratoriais são discordantes para um diagnóstico de traço de talassemia beta, é útil considerar outras condições que afetam o nível de Hb A2 e os índices de eritrócitos. Neste caso, uma importante pista foi encontrada na história clínica do paciente.
Hb níveis A2 tendem a aumentar em condições que retardam a maturação nuclear dos precursores de eritrócitos. Estas condições também estão associadas ao aumento do MCV.1,2 A causa mais comum deste fenômeno é a anemia megaloblástica devido à deficiência de folato e/ou vitamina B12. Entretanto, vários medicamentos que inibem a síntese de ácido nucléico têm efeito semelhante, incluindo a classe de medicamentos anti-HIV chamada inibidores da transcriptase reversa de nucleosídeos (NRTIs). A lamivudina no regime HAART deste paciente é um medicamento NRTI.
Então devemos concluir que o nível elevado de Hb A2 deste paciente foi devido à medicação anti-HIV, e o paciente não tinha traço de talassemia beta? Não tão rápido! Em geral, os aumentos na Hb A2 devido à NRTI (ou anemia megaloblástica) são menores que os observados no traço beta talassemia.1,2 Além disso, os aumentos tendem a ser proporcionais aos efeitos globais da droga, que podem ser aproximados por aumentos no MCV.1,2 Em muitos pacientes em terapia NRTI, o VMC é marcadamente alto (muitas vezes superior a 120 fL), enquanto que no nosso paciente, o VMC estava no extremo inferior da faixa de referência.
Por isso, não estávamos convencidos de que o nível muito alto de Hb A2 do paciente fosse devido apenas à terapia com lamivudina. Suspeitávamos que o paciente tinha condições concorrentes que aumentavam e diminuíam o VMC (ou seja, traço de talassemia beta e terapia com lamivudina, respectivamente), contribuindo ambos para um nível elevado de Hb A2. Relatamos ao médico solicitante que o traço de talassemia beta era provável e poderia ser confirmado pela análise da mutação da globina beta. O sequenciamento genético subsequente revelou uma mutação heterozigótica da beta globina associada à talassemia beta-zero.
Caso 2: fator de etnicidade
A paciente era uma mulher vietnamita grávida de 35 anos de idade que estava sendo rastreada para hemoglobinopatia. Os resultados laboratoriais são apresentados na Tabela 2.
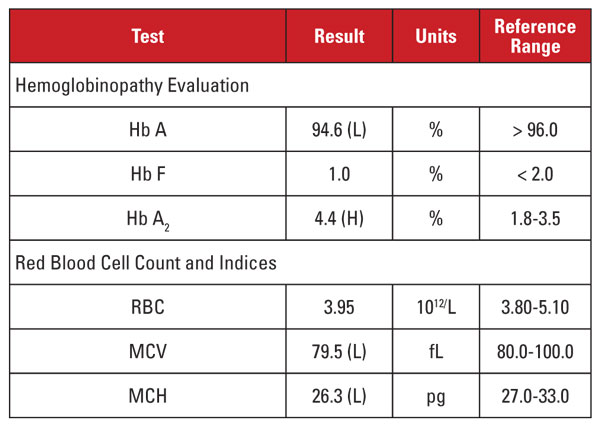
Again, o nível alto de Hb A2 foi discordante com microcitose e hipocromia mínima ou ausente. Neste caso, pudemos descartar fatores de confusão comuns, como folato, deficiência de vitamina B12 ou de ferro, disfunção tireoidiana ou efeitos medicinais. Com base na etnia do paciente, suspeitamos de outra importante causa de discórdia: o traço de talassemia beta co-herdou com a talassemia alfa menor. Quando um paciente herda ambas as condições, o MCV e a HGM frequentemente normalizam porque as cadeias alfa e betaglobina estão presentes em quantidades relativamente equilibradas no desenvolvimento de glóbulos vermelhos.2
Recomendamos análises genéticas para testar as mutações alfa e beta talassemia, a fim de avaliar completamente o risco para a mãe grávida e para o feto. A sequência subseqüente de beta globina revelou uma mutação heterozigótica beta-plus talassemia, enquanto a análise para deleções de alfa globina revelou uma deleção heterozigótica do sudeste asiático (SEA) de dois genes, consistente com -/αα alpha thalassemia minor. Com base nesses achados, a análise genética do pai foi indicada para avaliar o risco ao feto de herdar uma síndrome de talassemia clinicamente grave.
Talassemias alfa e beta
Talassemias estão entre as doenças hereditárias mais comuns na humanidade. São altamente prevalentes onde a malária tem sido endêmica, e agora são comuns em todas as partes do mundo devido à migração das populações humanas. As talassemias são causadas por mutações que reduzem a expressão do gene da globina nos precursores dos glóbulos vermelhos. As perturbações são classificadas pelos genes da globina que são mutantes (por exemplo, alfa versus beta) e pela gravidade da doença, que está relacionada com o facto de as mutações serem herdadas de uma forma heterozigota ou homozigota/composta heterozigota. A principal forma adulta de hemoglobina é a Hb A, um tetrâmero de duas cadeias de globina alfa e duas cadeias de globina beta. Na talassemia alfa, a expressão da globina alfa é deficiente e há um excesso correspondente de cadeias de globina beta.
Este padrão é invertido na talassemia beta. No cenário de deficiência e desequilíbrio da cadeia da globina, o conteúdo de hemoglobina nos glóbulos vermelhos é diminuído, resultando em microcitose e hipocromia. Além disso, o excesso de cadeias alfa ou beta globina formam tetrâmeros instáveis que causam hemólise. A talassemia alfa e beta distingue-se pela quantidade de hemoglobina Hb A2 adulta menor, um tetrâmero de duas cadeias de globina alfa e duas cadeias de globina delta. A Hb A2 está aumentada na talassemia beta porque a relativa falta de betaglobina permite a incorporação de mais cadeias delta na hemoglobina.
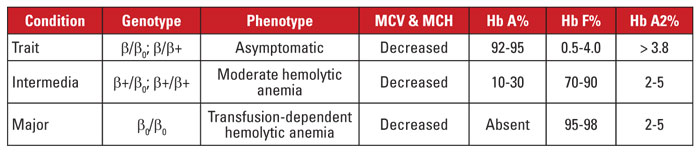
Beta talassemia é causada por mutações no locus do gene betaglobina no cromossomo 11,3,4 A maioria das mutações são pequenas substituições, inserções ou deleções de nucleotídeos, embora grandes deleções sejam identificadas em casos raros. Dependendo da mutação, a expressão da beta globina é reduzida parcialmente (beta-plus talassemia) ou completamente (beta-zero talassemia). A característica talassemia beta é causada por uma mutação heterozigótica. Esta condição é assintomática e caracteriza-se por aumento da Hb A2, microcitose de eritrócitos, e nenhuma anemia hemolítica significativa. Em contraste, a talassemia beta maior (anemia de Cooley) é causada por mutações homozigotos beta-zero. A avaliação da hemoglobina revela uma predominância de Hb F, ausência de Hb A, e Hb A2 normal ou aumentada. Beta talassemia major é caracterizada por anemia hemolítica grave, dependente de transfusão, com consequente esplenomegalia e deformidades ósseas. A transfusão recorrente leva à sobrecarga de ferro, que é uma das principais causas de morbidade e mortalidade. Beta talassemia intermedia é clinicamente heterogênea, com a maioria dos sintomas relacionados à anemia hemolítica moderada. A dependência transfusional é incomum porque a expressão da beta globina não está ausente. Devido ao grande número de mutações associadas à talassemia beta, o diagnóstico genético requer tipicamente o sequenciamento genético. As síndromes de talassemia beta estão resumidas na Tabela 3.
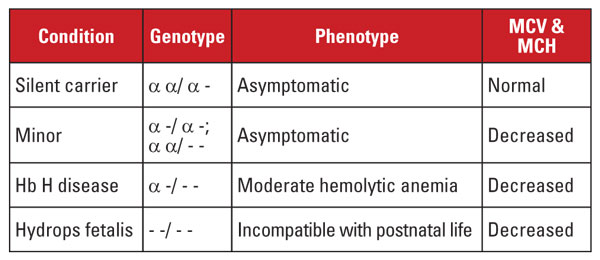
Talassemia alfa é causada por mutações no locus do gene alfa globina no cromossomo 16.5 Os genes alfa globina são duplicados no cromossomo 16, portanto, um indivíduo normal tem quatro cópias. As mutações mais comuns são as grandes deleções que afetam um ou ambos os genes no locus. Um número bastante limitado de deleções tem sido descrito, com prevalência em diferentes populações; por exemplo, grandes mutações que deletam ambos os genes alfa são muito comuns em pessoas da etnia do sudeste asiático. Essas mutações também são prevalentes em populações do Mediterrâneo, mas são raras em pessoas de etnia africana. Em contraste, as supressões de genes alfa globina simples têm uma distribuição generalizada. A perda de um dos quatro genes da globina alfa é referida como um estado portador silencioso.
Esta condição é assintomática e está geralmente associada com índices normais de glóbulos vermelhos. A perda de dois genes da globina alfa é chamada de talassemia alfa menor. Esta condição pode ocorrer devido a deleções homozigotos de um gene ou heterozigotos de dois genes. A contagem de células vermelhas e os índices são indistinguíveis do traço de talassemia beta, mas o nível de Hb A2 é normal. A perda de três genes alfa é chamada de doença de Hb H. (O Hb H é um tetrâmero das cadeias beta globina). A doença do Hb H é tipicamente uma anemia hemolítica moderada associada a esplenomegalia e alterações ósseas. A perda dos quatro genes alfa é denominada Hb Barts hydrops fetalis (Hb Barts é um tetrâmero das cadeias de gamaglobulina fetal). Esta condição é incompatível com a vida pós-natal. A Hb H e Hb Barts podem ser identificadas nas avaliações de hemoglobina e desempenham um papel no diagnóstico pós-natal de talassemia alfa.5
O diagnóstico genético é geralmente realizado por ensaios de reacção em cadeia da polimerase em fenda (PCR) que visam as deleções comuns. O sequenciamento do gene da globina alfa também está disponível para detectar mutações raras. As síndromes de talassemia alfa estão resumidas na Tabela 4.
Quando testes genéticos são necessários
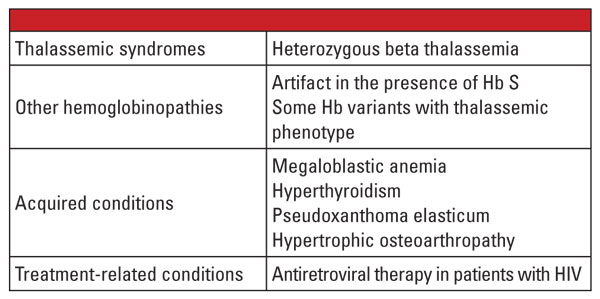
Em conclusão, as formas heterozigotas da talassemia alfa e beta são muito comuns e são identificadas freqüentemente no laboratório clínico. A avaliação da hemoglobina e outros dados clínicos e laboratoriais de rotina são geralmente suficientes para o diagnóstico. Os testes genéticos não são necessários na maioria dos casos, mas podem ser criticamente importantes no contexto do planejamento familiar. Se forem identificadas mutações de talassemia em ambos os pais, pode haver risco significativo para o feto de herdar talassemia grave. No caso de hidropisia fetal, é provável a morte do feto e há risco significativo para a mãe por complicações da gravidez.5 Portanto, é importante estar atento a variáveis que complicam o diagnóstico laboratorial das talassemias, e reconhecer quando os testes genéticos confirmatórios são indicados. Por exemplo, como mostrado no primeiro relato de caso, o diagnóstico do traço de talassemia beta pode ser desafiador se houver condições coexistentes que afetem o nível de Hb A2. Os fatores que são conhecidos por aumentar a Hb A2 estão listados na Tabela 5.6
- Steinberg MH, Forget BG, Higgs DR, et al. Distúrbios da Hemoglobina: Genética, Fisiopatologia Gerenciamento Clínico. Cambridge: Cambridge
University Press. 2009. - Steinberg MH, Rodgers GP. HbA2: biologia, relevância clínica e um possível alvo para a melhora da anemia falciforme. Br J Haematol. 2015;170(6):781-787.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76.
- Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619.
- Galanello R, Cao A. Gene revisão do teste. Alpha-thalassemia. Genet Med. 2011;13(2):83-88.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH recommendations for the measurement of haemoglobin A2. Int J Hematol de Laboratório. 2012;34(1):1-13.